Mr Justice Meade:
Introduction. 6
Case management 7
The issues. 9
EP949. 9
EP565. 10
Relief, the pledge issues. 11
The witnesses. 11
EP949 witnesses. 12
EP949 - Moderna's Expert, Professor Rosenecker 12
EP949 - Pfizer/BioNTech's expert, Dr Enright 13
EP565 witnesses. 17
EP565 - Moderna's expert, Dr Ulmer 17
EP565 - Moderna's expert, Dr Sola. 20
EP565 - Pfizer/BioNTech's expert, Prof Dougan. 21
EP565 - Pfizer/BioNTech's expert, Prof Weiss. 22
EP565 - Pfizer/BioNTech's expert, Prof Alabi 23
Pfizer/BioNTech witnesses - general 24
The skilled person - the law.. 24
Added matter and novelty - the law.. 29
The basic tests. 29
Individualised description and selection from lists. 31
Selection from multiple lists. 34
Obviousness - the law.. 37
EP 949 - The common general knowledge. 40
Agreed CGK.. 40
DNA.. 40
RNA.. 43
Transcription. 46
mRNA Processing. 46
In vitro transcription. 47
Translation. 48
Modified Nucleotides. 48
Applications of RNA in research and therapeutics. 50
IVT mRNA as a potential therapeutic agent 52
Approaches to improving IVT mRNA.. 53
Transfection/delivery. 53
Improved 5' caps. 53
Polyadenylation. 53
Untranslated regions. 54
Reducing Immunogenicity. 54
Codon optimisation. 54
Nucleic acids and the immune system.. 54
Innate immunity. 54
Pattern recognition receptors. 54
Nucleic acids and innate immunity. 55
Karikó 2005. 56
Disputed CGK.. 56
Kormann 2011. 57
EP949 - The Skilled Person. 57
The EP949 specification. 60
Examples. 63
Claims in issue. 64
Validity - EP949. 65
Disclosure of UPenn. 65
Example 2. 67
Example 7. 68
Example 31. 69
Novelty of EP949 over UPenn. 70
Route 1. 71
Route 2. 71
Moderna's response to Routes 1 and 2. 72
Route 3. 74
Decisions of other jurisdictions on EP949. 75
EPO Proceedings. 75
The decision of the Court of the Hague. 76
EP949 Obviousness analysis. 79
Charette & Gray. 84
Experiments with unpredictable results. 90
One way street 90
The EPO technical contribution case. 91
Secondary evidence. 91
Similarities and differences between UPenn and Karikó 2008. 93
Insufficiency - EP949. 96
EP565 Introduction. 96
EP565 skilled team.. 96
EP565 - The common general knowledge. 99
Agreed CGK.. 99
Coronaviruses - Overview.. 99
Classification of coronaviruses. 99
Coronavirus structure and genome. 100
Spike Protein (S) 101
Diseases caused by human betacoronaviruses. 102
The 2002-2003 SARS outbreaks. 102
MERS outbreaks. 102
SARS-CoV and MERS-CoV neutralising antibodies and animal models. 102
The Immune System.. 104
Vaccines - Overview.. 104
Vaccination. 105
Types of vaccine. 105
Vaccine Design. 106
Evaluating vaccines. 107
Nucleic Acids. 109
Nucleic acid vaccines. 109
DNA Vaccines. 110
RNA vaccines. 111
Non-self-amplifying/conventional mRNA optimisation. 111
Self-amplifying mRNA vs non-self-amplifying/conventional mRNA vaccines. 112
RNA vaccine targets. 113
Vaccine associated enhancement of disease. 113
Coronavirus Vaccine Development at the EP565 Priority Date. 114
Delivery of Nucleic Acids. 114
Design of delivery systems for nucleic acids. 115
Lipid-based carriers for nucleic acid delivery. 116
Lipoplexes. 116
Liposomes. 116
Lipid nanoparticles (LNPs) 117
Cationic nanoemulsions. 117
Disputed CGK.. 118
Issue (a): Whether betacoronaviruses were a vaccine target at the EP565 Priority Date 118
Geall 2012. 123
Issue (e): The skilled team's view as to the relevant factors for an antigen-specific immune response by a nucleic acid vaccine. 124
The EP565 specification. 125
The reference examples (examples 12 to 19) 128
The betacoronavirus examples (examples 20 to 24) 129
Claims in issue. 132
EP565 Claim interpretation. 132
Disclosure of WO674. 133
Examples. 134
Disclosure of Pardi 136
EP565 novelty and added matter 137
Adequate disclosure of the physical features in the application as filed (added matter) 140
The Opposition Division Decision. 142
Plausibility as part of the added matter argument 142
Adequate disclosure of the physical features in WO674 (novelty) 145
EP565 obviousness. 146
Assessment 147
Pardi 151
EP565 dependent and proposed amended claims. 151
Insufficiency - EP565. 152
Conclusions. 152
1. This is the trial of two actions, HP-2022-000022 and HP-2022-000027, concerning the following pair of European Patents (collectively "the Patents" or sometimes in the context of just one "the Patent"):
i) European Patent (UK) No. 3 590 949 ("EP949"); and
ii) European Patent (UK) No. 3 718 565 ("EP565").
2. For convenience, and because this trial focused primarily on revocation rather than infringement, I will where appropriate use the terms 'Claimants' and 'Defendant' to refer to the designations in the revocation action (HP-2022-000027). More usually I will refer to the Claimants as "Pfizer" and "BioNTech", and together as "Pfizer/BioNTech".
3. The Patents are in the name of the Defendant ("Moderna"). Both patents are asserted against Pfizer/BioNTech's SARS-CoV-2 vaccines.
4. Infringement was not in issue; effectively it was admitted, with the Claimants saying that they did not dispute infringement on any interpretation of the claims advanced by either side. There was no issue of fact about the alleged infringing products.
5. EP949 is entitled "Ribonucleic acids containing N1-methyl-pseudouracils and uses thereof" and concerns messenger RNA ("mRNA"). It claims mRNA in which one of the usual nucleosides (uridine) is replaced with N1-methyl-pseudouridine ("m1Ψ"). EP949 has a priority date of 1 October 2010 (the "EP949 Priority Date"), which was not challenged in these proceedings. The prior art primarily concerned pseudouridine ("Ψ"), although the main citation mentions m1Ψ. The structures of Ψ and m1Ψ are shown at paragraph 306 below.
6. EP565 is entitled "Respiratory virus vaccines" and relates to a betacoronavirus mRNA vaccine formulated in a lipid nanoparticle, and the use of such an mRNA vaccine in a method of preventing and/or treating betacoronavirus disease. Moderna relied on the ninth priority document, dated 28 October 2015 (the "EP565 Priority Date"), but priority was challenged by Pfizer/BioNTech and Moderna conceded it shortly after opening skeletons were exchanged. The filing date of EP565's application is 21 October 2016 (the "EP565 Filing Date") and that is therefore the date for assessing its validity.
7. Moderna is a pharmaceutical company based in the US. It focuses on exploring potential uses for mRNA in medicine. Pfizer Inc. is a pharmaceutical and biotechnology company based in the US, and BioNTech SE is a biotechnology company based in Germany. In March 2020, BioNTech and Pfizer announced their partnership for the development, testing, manufacturing, distribution and regulatory approval of an mRNA-based SARS-CoV-2 vaccine. This vaccine is now known as Comirnaty.
8. At trial:
i) Piers Acland KC represented Moderna on EP949, leading Stuart Baran;
ii) Andrew Waugh KC dealt with EP565 for Moderna, leading Katherine Moggridge and Richard Darby;
iii) Tom Mitcheson KC conducted the oral advocacy for Pfizer/BioNTech on EP949, with the exception of closing submissions relating to secondary evidence on obviousness, which were dealt with by Ms Hart;
iv) Michael Tappin KC was the advocate at trial for Pfizer/BioNTech on EP565, leading Michael Conway;
save in relation to some legal issues on added matter and novelty which were common to both actions, as explained below, where leading Counsel crossed over somewhat.
9. I am grateful that regard was had by Pfizer/BioNTech for the encouragement in the Patents Court Guide (and the recent speech of the Lady Chief Justice) for parties to make greater use of junior advocates. However, it was an equally valid choice to have the same advocate deal with all relevant issues for each party on each Patent.
10. Pfizer and BioNTech had separate solicitors and, as I understand it, formally speaking their respective Counsel were only instructed for Pfizer (Mitcheson KC/Hart) or for BioNTech (Tappin KC/Conway). What I have just outlined concerning the trial describes what they covered in Court. Where I refer below to "Counsel for Pfizer/BioNTech" it should be understood in that context.
11. There are parallel proceedings in a number of jurisdictions, including the USA, the Netherlands, Germany, Belgium, Ireland and the EPO. Various of the proceedings are stayed pending the EPO or PTAB review. There have been first instance decisions in the Netherlands and the EPO, to which I refer below.
12. All these proceedings were, of course, brought against the background of the Covid-19 pandemic, which has meant that the amounts at stake are very large. But I must bear in mind that at the dates of both Patents the events of the pandemic with their profound and global effects were still far in the future and can have no significance whatsoever to validity of the Patents. Both sides were guilty of using the events of the Covid-19 pandemic by way of illegitimate hindsight and I will ignore that (on EP565 I heard evidence about concerns at the EP565 Priority Date as to future coronavirus outbreaks, that is a different matter and I deal with it below). Similarly, the fact that authors of the prior art on EP949 later won the Nobel Prize, in 2023, is irrelevant, although they were already eminent at the EP949 Priority Date, and the reader of the prior art would have known that.
13. This trial was effectively two distinct patent actions heard at the same time. EP949 and EP565 have significantly different priority dates; there was little to nothing in common on the CGK; the experts were different; the Counsel teams were different; on the Pfizer/BioNTech side the solicitors were different.
14. There is nothing wrong with hearing actions on two separate patents in the same trial and where it is necessary or helpful to resolving the real commercial dispute between parties, then of course the Patents Court will consider doing it (within reason - in telecoms cases there may be more patents in issue and there are limits to how many can be done in one trial, but equally the commercial dispute often does not depend on any individual patent). Indeed, trying everything at issue between the parties at once is in a sense the default. But it does impose a significant burden, as I will explain, and there are lessons to be learnt from the way this trial was run.
15. The first issue is that melding two trials into one in this way presents timing issues, and timing decisions to be made. In particular: should all the opening submissions come at the start on all the patents, and then all the evidence and then all the closings? Or should the two patents be scheduled as if two separate trials, back to back?
16. This is of course a case management decision for the Court, to be taken in cooperation with the parties, and there are things to be said in favour of either course. In the present case, at the PTR the parties had two possible timetables to suggest, not that different from each other, but neither was at all optimal. The one I selected from the parties' proposals put EP949 openings and evidence first, then EP565 openings and evidence, then all the closings. This was no doubt fine for the parties and especially the EP949 Counsel who had plentiful time to work on their closings with no distractions, but it was neither fish nor fowl, neither one big trial nor two trials back to back, and meant I had to hear the EP949 closings after a number of days and almost the whole of the EP565 trial had unfolded in the time since the EP949 evidence (ironically, at one point Moderna complained about one of my more minor timing suggestions because it would have meant one of its solicitors having to work on both Patents during a particular period).
17. At the PTR, I pointed out this then-potential problem but by then the parties had made practical arrangements around the limited options they had identified and I decided not to disrupt them by steering my own course. In future, in multi-patent trials parties should not sell the pass like this, and if they feel they need to make structural decisions about the organisation of the trial in advance of the PTR, they must engage with the Court about it as early as possible and without committing to anything.
18. The second issue is that if the parties decide to have separate legal teams within the same multi-patent trial (it has become common also to do so even in single-patent cases), and if part of the reason is that the cases are seen to be discrete, the parties must still keep an eye on whether there is overlap, and must actively manage it if there is.
19. At this trial, the added matter/novelty issues between EP949 and EP565 overlapped a lot in terms of the applicable law, and although the facts were entirely different, it might be argued that (by way of abstract example) if X applied to novelty on EP949 then by parity of treatment Y ought to apply on added matter on EP565 (a dynamic which was further complicated, I suspect, by whether winning on one patent and losing on the other would be an overall victory for either party). But this overlap seemed to go unremarked by the parties until I raised it when I noticed that Moderna was arguing in favour of the EPO "serious contemplation" standard on EP565 added matter, and against it on EP949 novelty. This had to be addressed by my requiring the parties to put in single sets of submissions on the law and rearranging the closing oral submissions to put all the added matter and novelty law together, whereupon a number of the overlaps and inconsistencies were resolved. Counsel for Pfizer/BioNTech said that the tensions and inconsistencies arose much more keenly on Moderna's side, and there is some truth in that, but it was not wholly one-sided, and both parties should have spotted the possibility to avoid a waste of time and fruitless increase in complexity.
20. The third issue is that having two legal teams on each side seems to hinder the parties from reducing the issues to focus on what is important. Issues did drop away in this case, most notably when Moderna gave up on priority entitlement for EP565, but not to the extent I would have liked to see, and not to the extent that I think would have been possible. For example, some dependent claims remained in issue, or at least their status remained unclear, longer than was necessary, and on both Patents Pfizer/BioNTech kept their second-string prior art in play without sufficiently considering whether it was justified. I recognise that the amounts of money at stake in this dispute might justify keeping points going from a purely financial, cost:benefit perspective, but parties still must focus on what is important. When there are two separate legal teams engaged over the whole of a multi-patent trial, their greater bandwidth (greater than one team, certainly greater than one judge) is bound to reduce the practical impetus to simplify.
21. The issues for EP949 are:
i) The identity of the skilled person;
ii) The scope of the CGK, although the dispute dwindled to nothing, or almost nothing, in the course of closing arguments;
iii) Anticipation by International Patent Application WO 2007/024708 A2 ("UPenn");
iv) Obviousness over:
a) UPenn; or
b) Karikó et al, "Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability" Molecular Therapy 2008; 16(11):1833-1840 ("Karikó 2008"); and
v) Insufficiency as an enablement squeeze expressed in the following form: the disclosure of EP949 is no more enabling than that of the common general knowledge and prior art. This did not really feature much at all at trial.
22. Pleaded issues of added matter and insufficiency against claim 4 fell away after Moderna decided not to maintain the independent validity of claim 4 at trial.
23. At the end of the oral evidence I asked Counsel for Pfizer/BioNTech in what scenario obviousness over Karikó 2008 could succeed if the case over UPenn failed. Pfizer/BioNTech felt unable to concede that Karikó 2008 could not succeed if UPenn fell short, although they framed their closing submissions primarily by reference to UPenn and kept Karikó 2008 really only out of caution. Both sides identified certain differences. The attacks are extremely similar, though, and the contents of the two documents, which are from the same team, overlap a great deal. So I will address UPenn first, then explain fairly briefly the differences from Karikó 2008 and why they do not lead to any different result.
24. The issues for EP565 are:
i) The identity of the skilled person;
ii) Two disputes over CGK;
iii) Anticipation by and obviousness over a Moderna application WO 2015/164674 ("WO674");
iv) Obviousness over Pardi et al, "Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes" Journal of Controlled Release; 217(2015): 345-351 ("Pardi"); and
v) Added matter.
25. A number of issues fell away shortly before the trial began:
i) Anticipation by and obviousness over a Novartis application (WO 2012/006369 A2) were dropped by Pfizer/BioNTech ahead of the exchange of skeleton arguments;
ii) A dispute as to whether EP565 is entitled to a priority date of 28 October 2015 fell away after Moderna decided not to seek to maintain priority;
iii) Obviousness over a Novartis application (WO 2015/095340 A1) fell away after Pfizer/BioNTech decided not to rely on it at trial. This followed Moderna's decision not to defend priority entitlement; and
iv) Additional pleaded insufficiency and AgrEvo obviousness attacks also fell away in the lead up to trial (not in the sense of being abandoned but because Pfizer/BioNTech said they had done their job and did not need argument or separate resolution).
26. Moderna has both conditional and unconditional applications to amend the claims of EP565. Pfizer/BioNTech opposed the unconditional amendments on the basis that they do not cure invalidity.
27. There were a variety of dependent claims, both as unconditionally and conditionally amended, but in the end the ones that mattered were claims 1 and 10. I sweep up the other claims including the various amendments in a separate section.
28. Moderna does not seek injunctive relief in these proceedings, only financial remedies.
29. During the Covid-19 pandemic, Moderna made public statements that (I paraphrase for simplicity) it would not assert its patent rights while the pandemic continued. In due course, it stated its belief that it was entitled to, and would, begin to assert its patent rights again, to seek financial compensation.
30. In these proceedings, Pfizer/BioNTech have relied on Moderna's statements as at least a partial defence to financial remedies. These were referred to as the "pledge" issues. Earlier in the proceedings I made a case management order that the pledge issues would be determined in a separate trial before another judge. Jonathan Richards J accordingly heard the pledge issues at a trial that took place simultaneously with this one.
31. Moderna called three experts and Pfizer/BioNTech called four experts. They were in the fields of:
i) Nucleic acid biology (EP949) (the notional person in this discipline was sometimes referred to by the more specific shorthand "RNA biologist");
ii) Nucleic acid vaccinology (EP565) (the abbreviation "NAV" was used for "nucleic acid vaccinologist");
iii) Coronavirus virology (EP565) ("Coronavirus Virologist"); and
iv) Lipid nanoparticle ("LNP") formulation and manufacture (EP565).
32. There were disputes about the boundaries and relationships of these fields and whether and to what extent these experts would be part of the skilled team. I have adopted short labels for them here for simplicity and without prejudging any of those issues.
33. Quite a few of the parties' criticisms of the other side's experts depend on points of detail, some of them complex. I have preferred nonetheless to deal with them all together here, at the start of the judgment so that their totality can be appreciated. I realise that they may be harder for the reader to follow than if I dealt with them in context but prefer this course to scattering them widely throughout the judgment. A reader who wants to track the logic of the points carefully but who does not already know the detail of the case may wish to read the rest of the judgment and then return to this section.
34. Moderna relied on evidence from Professor Josef Rosenecker (RNA biologist). Pfizer/BioNTech relied on evidence from Dr Anton Enright (RNA biologist). Moderna had also filed an expert report in chief from Professor Clare Bryant (immunology), but this was withdrawn after it was established between the parties at the PTR that there would be no specialist innate immunologist member of the skilled team.
35. No fact witnesses were called.
36. I am going to deal with Moderna's witnesses first in relation to each patent. This does not reflect the order of giving evidence but overall is more convenient.
37. Between 1988 and 2003 Professor Rosenecker's research focused primarily on DNA-based gene therapy of lung diseases. Much of his work centred on treating patients with cystic fibrosis. From 2003 he began working on RNA, with a goal of using small interfering RNAs (siRNAs) as a way of suppressing expression of the epithelial Na+ channel, which was thought to be hyperactive in patients with cystic fibrosis.
38. Professor Rosenecker became interested in using mRNA to express transgenes in the mid-2000s. In 2007 he received a grant as coordinator of a number of researchers investigating mRNA transfer for treating cystic fibrosis.
39. Since the Covid pandemic, Professor Rosenecker's group has been involved in the development of mucosal mRNA vaccines.
40. Professor Rosenecker gave his evidence in English (both orally and in his reports), despite it not being his first language. My clear impression was that he is fluent in English and fully understood what was being said, but nonetheless some of his answers in oral evidence read in a somewhat awkward way on the page. I have borne this in mind; obviously it was in no way his fault.
41. One consequence of Prof Rosenecker giving evidence in English was that he found the experience more tiring. Again, this is entirely understandable and not his fault, and on one occasion he very sensibly asked for the Court day to finish early (to which I agreed) because he felt his concentration had dropped too much. I noticed the drop in his concentration myself and take it into account in assessing the evidence he was giving in the immediate period beforehand.
42. In their closing submissions, Pfizer/BioNTech criticised Professor Rosenecker for occasionally attempting to avoid answering questions and preferring to answer something else, particularly "when it came to answering some of the crunch questions on obviousness". Pfizer/BioNTech would of course have preferred an answer more in keeping with their case, but in my view he was answering in an honest, direct way and stating reservations which he genuinely held.
43. Pfizer/BioNTech submitted that because Professor Rosenecker was not involved in the RNA biology aspects of the research his team conducted on gene therapy, he was not the right member of the team to be giving evidence on the relevant issues in the case, including the matter of modified nucleoside selection. Pfizer/BioNTech pointed to the fact that despite being involved in an mRNA vaccine project, Prof Rosenecker was unaware that m1Ψ was used in the vaccines distributed during the Covid-19 pandemic. In my view, while it is true that Dr Kormann, a member of Prof Rosenecker's team had more specific knowledge of modified nucleosides and hence made the detailed decisions about which ones to use in the group's work, it is a non sequitur to say that Prof Rosenecker did not have enough knowledge to give the evidence that he did and to assist me. He obviously did, and spoke with knowledge and authority about, for example, the detailed characteristics of how modifications to Ψ could affect activity, the effect of changes in the Watson-Crick interface, and so on.
44. Pfizer/BioNTech also made a submission regarding the narrowness of the skilled team Professor Rosenecker had been instructed to consider, and I address this in the skilled person section below.
45. Pfizer/BioNTech sought to make an issue of the fact that Prof Rosenecker used the expression "transcript therapy" and said that this was not a phrase in use as a term of art at the EP949 Priority Date, indicating, they said, that there was no such distinct field. It also criticised Prof Rosenecker personally over his usage. I do not think there was anything in this. I accept that Prof Rosenecker did use the term himself, and it is a term which, among RNA biologists of the general kind under consideration, is self-explanatory even to someone who might not have heard it before.
46. Finally, Pfizer/BioNTech said that Prof Rosenecker's evidence as a whole was undermined by his taking a position on the CGK status of the RNA Modification Database ("RNAMD") from which he had to retreat and on which issue Moderna in due course gave up altogether. My conclusion on that point is that Prof Rosenecker was out of step with the notional skilled person in being somewhat less technically savvy and used to online resources, and that did lead him into a degree of inconsistency on this point, including inconsistency with previous statements of his own. But it went no wider than that and did not reflect on his reliability generally.
47. Pfizer/BioNTech's expert was Dr Anton Enright (RNA biologist).
48. Dr Enright is currently University Associate Professor, Group Leader and Academic Lead of Genomics in the Department of Pathology at the University of Cambridge.
49. Before his current role, Dr Enright was a Junior Investigator on the Genome Campus at the Wellcome Sanger Institute in Cambridge from 2004 and became the Research Group Leader at the European Molecular Biology Laboratory European Bioinformatics Institute in 2009.
50. Moderna made the following submissions about Dr Enright's experience and evidence:
i) First, in relation to his experience:
a) That he had no direct experience of using mRNA for therapeutic purposes or experience with attempting to increase expression of exogenous mRNA;
b) that his interests related to types of RNA which are not translated (microRNAs, piwi-RNAs and long non-coding RNAs); and
c) that his interests lay in matters of fundamental research and fundamental biology.
ii) Second, that Dr Enright knew that the case related to m1Ψ at the time of his first report and that parts of his evidence were contaminated by hindsight.
iii) Third, that Dr Enright's evidence was inconsistent in material aspects. Moderna cited Dr Enright's evidence on the skilled person's reliance on the RNAMD and the skilled person's interest in learning more about the properties of Ψ.
iv) Fourth, that Dr Enright's approach to obviousness was erroneous due to his willingness to entertain modifications which could produce negative ("catastrophic") results.
51. As to the first point, it would be unfair and inaccurate to say that Dr Enright's work was purely abstract or computational (although computational analysis was a very strong feature of his work over some periods) and he plainly had considerable experience of "wet" laboratory work. But I do accept that his interests were to do with fundamental research, and well removed from the practical application of mRNA expression, whether for therapeutic or any other applied goals. He pointed out that all work in his field was done with the general goal, ultimately, of improving human health, but that does not change the fact that his work was much more at the theoretical end of the spectrum. As a result, he was given to thinking that things were obvious to do if they would yield information of any kind, positive or negative, and whether or not they would give a practical advantage. The fourth point above is a facet of this, and I accept it.
52. The second point concerns the risk of hindsight and I need to spend a little time on it.
53. Dr Enright explained in his first report that he was asked for his views on the CGK and prior art before he was shown EP949, and he also said that he did not know the modified nucleotides used in Covid-19 vaccines at the initial stage. Naturally, as Counsel for Pfizer/BioNTech accepted, by the time he finalised his first report he knew that the invention of EP949 was about m1Ψ, and there is nothing wrong with that.
54. Counsel for Moderna did not directly probe or challenge the process of Dr Enright's instruction and I therefore accept that in general it followed the desirable "sequential unmasking" approach that has been discussed in a number of judgments in patent cases.
55. However, that does not mean that all the main elements or details of Dr Enright's reports and the analysis leading to his conclusion of obviousness were arrived at before he knew about m1Ψ, and Moderna submitted that there was objective reason to think that there was at least some hindsight at play. It relied on two examples.
56. The first relates to his treatment of [0056] of UPenn. As is discussed below, in that paragraph, the authors refer to the four naturally occurring Ψ derivatives known at the time, plus m5D. These are defined as being pseudouridines.
57. An oddity, however, is that m5D is not a pseudouridine derivative like the others mentioned. It is common ground that the skilled person would think about this and wonder why it was listed. In paragraph 7.16 of his first report, Dr Enright said "The Skilled Person would note that while m5D is not literally a Ψ derivative like the others in the group, it does share structural similarities with the others, in particular m1Ψ, which would explain its inclusion."
58. It is not in dispute that m5D is in fact particularly similar to m1Ψ, but Counsel for Moderna put to Dr Enright in cross-examination that he would not have offered the explanation that he did, in the way he did, unless he had an awareness of the importance of m1Ψ to this case. This was fortified by the fact that Prof Rosenecker, in his second report, pointed out that there are other nucleosides in the RNAMD not mentioned in [0056] which are more closely related to the other modifications in that paragraph than m5D is to m1Ψ. In particular, he pointed to there being just one difference between Ψm and Um.
59. Dr Enright agreed that the skilled person would do an analysis similar to Prof Rosenecker's but would not chase "down the rabbit hole" of every detail.
60. Counsel for Pfizer/BioNTech said that Dr Enright was doing no more in his paragraph 7.16 than stating a fact about similarity. I do not agree. If unaware of any particular importance of m1Ψ, Dr Enright could have made much the same point without using the words "in particular m1Ψ". He also plainly went further than just stating a fact because he was seeking to explain what the skilled person would think the patentee's logic was that underlay the inclusion of m5D. I think that without knowledge of the importance of m1Ψ, Dr Enright would have more likely gone on to say something along the lines of there being a potential reason why m5D was included, but that it was obscure why it was the only nucleoside mentioned that was not literally a Ψ derivative, given that there were other derivatives even closer to those mentioned in [0056]. In other words, Dr Enright's express mention of m1Ψ when it was not entirely necessary to making his general point, and stopping the explanation on the basis of the comparison to m1Ψ when there was more to be said, are both indicative of a particular focus on m1Ψ not to be found in [0056] or UPenn generally, feeding into his obviousness analysis.
61. The second example related to a paper by Brand and others from 1978 Biochem J referenced in the RNAMD in the entry for m1Ψ, which Dr Enright relied on during his oral evidence as showing that m1Ψ was present in 18S RNA of certain HeLa cells. I agree with Moderna that Dr Enright's reliance on Brand in this way was inconsistent with his position as to the skilled person's attitude to Charette & Gray (which I deal with in more detail below and which is referenced much more prominently in relation to Ψ in the RNAMD), and also that the inconsistency is hard to explain without reference to hindsight. However, this point does not bear on the preparation of his written reports, only his oral evidence.
62. I also thought that Dr Enright's very heavy focus on [0056] in UPenn relative to [0291] is something that would have been unlikely to the reader of the document without hindsight.
63. For these reasons I conclude:
i) Based on the [0056]/paragraph 7.16 point, that while Dr Enright generally tried to prepare his written evidence analysing obviousness without knowledge of the importance of m1Ψ, that knowledge came in to at least some degree. It is not possible to be sure to what extent, but I think it was appreciable.
ii) Based on the Brand point, the emphasis on [0056] and his evidence generally, there was some material hindsight in Dr Enright's approach overall.
64. Neither of these is a personal criticism of Dr Enright. I accept his general evidence about the sequence of his instructions, but it is a reality of the system that his long and detailed report will have continued to be worked on after he did know about the importance of m1Ψ, and it is an understandable thing that it affected the analysis. As to there being some general hindsight, this is a factor to take into account and not a reason for rejecting his evidence wholesale.
65. As to the third point above, inconsistency, I do not think it was a general feature of Dr Enright's approach, even if occasionally there was an internal tension in what he was saying (for example with Brand, above).
66. Based primarily on the second and fourth points concerning Dr Enright, above, I consider that Prof Rosenecker was a more useful witness in helping me to understand how the skilled person would think and reason at the EP949 Priority Date. But both witnesses were helpful in assisting me to understand the technology involved.
67. Moderna relied on evidence from:
i) Dr Jeffrey Ulmer (NAV); and
ii) Dr Isabel Sola (Coronavirus Virologist).
68. Pfizer/BioNTech relied on evidence from:
i) Professor Gordan Dougan (NAV);
ii) Professor Susan Weiss (Coronavirus Virologist); and
iii) Professor Christopher Alabi (drug delivery chemist, specifically LNP formulation and manufacture).
69. No fact witnesses were called.
70. Dr Ulmer has worked in the field of nucleic acid vaccine research and development for over 30 years. His qualifications include a degree in chemistry, a PhD in biochemistry and a postdoctoral fellowship at Yale University School of Medicine. Between 1990 and 1998 he worked at Merck & Co on DNA vaccine programs, including work on the development of a DNA vaccine for influenza. In 1998 he moved to Chiron Corporation to lead its DNA vaccine technology group and in 2007 he became site head for US Vaccines Research at Novartis. At Novartis he focused on the development of self-amplifying saRNA platform technology. From 2008 to 2015 he was Global Head, External Research at Novartis. Dr Ulmer began working as an independent consultant in 2020 and he is currently president of TechImmune LLC, a company developing an mRNA LNP universal coronavirus vaccine. Counsel for Pfizer/BioNTech criticised Dr Ulmer on a number of fronts.
71. First, Pfizer/BioNTech said that he did not have the detailed knowledge of the manufacture, formulation and use of LNPs that the skilled team would need to implement the invention of EP565 and that the evidence of Prof Alabi should be preferred. I agree that Prof Alabi had much fuller information, knowledge and experience on aspects of these matters and Moderna did not really argue otherwise. Moderna's case in opposition to the obviousness case does not depend on arguing that the skilled team would think that they could not make a suitable LNP formulation starting from WO674 or Pardi, though, so this matters little. Rather, Dr Ulmer gave evidence about whether it was predictable that mRNA delivered via an LNP would result in a properly expressed immunogenic protein and I think he was adequately qualified to opine on this. Likewise, although there were flaws in his evidence, he was qualified to speak to coronaviruses as possible vaccine targets from the point of view of the NAV.
72. Second, Pfizer/BioNTech said that he lacked a broad overview of the vaccine field (compared to Prof Dougan) because he had spent his whole career at Chiron/Novartis/GSK. I reject this. There may have been points on which he did not have specific knowledge (for example about which vaccine developers were working on MERS) but he was plainly a person with wide industry exposure and understanding who participated actively in international meetings and discussion.
73. Third, it said that he was seeking to defend a position and argue Moderna's case. I did not detect this at all. He gave very concise answers that were responsive to the questions put, including in cases where the yes/no answer that he gave might well have helped Pfizer/BioNTech. I agree that an answer that he gave about why companies were working on MERS-CoV vaccines was a bit speculative, and the same applies to an answer about how the Korean outbreak in 2015 came about. But these minor points must be seen in the context of his extensive evidence. He was also criticised for questioning some positions taken by CEPI and not agreeing with Counsel for Pfizer/BioNTech on the implications of a statement by Dr Rappuoli made in a 2014 article, but I think those were genuine views and Dr Ulmer was under no obligation to uncritically accept everything said by even such well known figures, or the implications.
74. Fourth, Dr Ulmer gave a talk in 2014 at a vaccines conference, where the following question and answer took place:
Questioner 5: Couple of quickies Jeff, how long does the immune response last for? And can it be [...] Question 2 is, there's a bit of a sort of oxymoron to [...] you want the vaccine to be available rapidly, that's one thing, that's great, on the other hand if you have a cross protective vaccine of long-lasting immunity, why do you need a rapid response to a vaccine anyway, because you're protected?
Dr Ulmer: Well to answer your second question first, I mean it, that may be true for flu if we can come up with ways to better induce long-lived cross protective immunity then it would obviate the need for this, but that doesn't solve all the other newly emerging pathogens like MERS and SARS and Ebola, and your first question in terms of longevity in small animals, it's like most vaccines, it lasts a long time. In primates the immunity does wane as it does with many other kinds of vaccines but it's boostable, if you recall the primate data with HIV envelope, you could see that it rapidly induced responses during the priming phase which slowly decayed but could be boosted tenfold by follow up boosting.
75. When asked about this in cross-examination, the following exchange occurred:
Q. If we just go over to page 19 of the bundle, can I ask you to read to yourself what questioner 5 says and what your response was? (Pause for reading)
A. Yes, I have read it.
Q. So you have singled out MERS and SARS, along with Ebola; yes?
A. Yes, I have read this carefully and, unfortunately, the entire question is not there. Some of it seems to be inaudible and not transcribed, so in response to question 2 what the questioner is asking me is, I talked about both the induction of long lived cross-protective immunity, as well as a rapid response, and I think the questioner is asking why would you need both. If you have long-lasting protection, why would you need a rapid response? So I think that is how I interpret the question. So my answer is that for a disease like influenza where there is at least theoretically a possibility of inducing long-lived cross-protective immunity, because of our deep knowledge of the virus and the fact that there are conserved proteins in the virus that could be targeted for that purpose, that maybe you would not need to have both in that case. But in the case of an outbreak situation, where it is a newly emerging, a re-emerging pathogen, you would want to have the capabilities of a rapid response. My specific comment to MERS and SARS, I can see the comment to Ebola, that makes sense because I have been talking about Ebola, and it is possible I even showed some data on Ebola in this talk, because we had a collaboration with the US army research on Ebola. But the only reason I can think of that I specifically called out MERS and SARS is because that was brought up in the question, but I do not recall the question.
76. It is clear, having reviewed the video, that Dr Ulmer's reference to MERS and SARS was not triggered by the question from the audience, and Dr Ulmer, having watched the video in the witness box at the end of his cross-examination, quite readily agreed as much. However, Counsel for Pfizer/BioNTech put to him earlier in his oral evidence that he had in fact already watched the video (i.e. before his oral evidence) and therefore knew that the idea of MERS/SARS had not come from the audience at the time when he offered it as a possible explanation; in other words that he fabricated the explanation knowing it was false.
77. I reject this, although it was a fair topic for the cross-examiner to explore. I believe Dr Ulmer that he had not already watched the video when first asked, and that he was genuine in his initial belief that MERS/SARS were not on his radar at the time of the talk so the idea must have come from the questioner. Clearly, he was wrong about that, as I have said, and the result is that I must and do conclude that SARS/MERS were on Dr Ulmer's radar in 2014. The fact that he got this wrong is a failure of recollection, and an important one - it makes me very reluctant to accept his view of what NAVs in the field did and did not know about risks from coronaviruses then - but it does not mean that he was anything other than honest. I think he was entirely honest.
78. As a fallback Counsel for Pfizer/BioNTech argued that Dr Ulmer should have reviewed the video before he commented on what had happened and why. I agree that if he had done so then his mistake would have been avoided, but there was, I am sure, a lot for him to do in preparing to give evidence, not least review a considerable amount of paper in the CXX bundle and I do not think it is a material criticism of him that he did not watch the video. He was, in general, extremely careful about his evidence.
79. Fifth, a number of instances were identified where it was said that Dr Ulmer had made a bad point, or a point which he could not defend in the witness box. There were some of these, but I did not think they went beyond making the odd mistake.
80. Sixth, and most importantly, Counsel for Pfizer/BioNTech said that Dr Ulmer's overall approach reflected a degree of pessimism not representative of the skilled team when it came to assessing prospects of success, and, relatedly, a view on what amounts to an inventive step quite different from the standards applied in these Courts. I think this point was well made and well supported and I accept it. I do not think these were attitudes that Dr Ulmer adopted for the purposes of his evidence, and I do not think they were synthetic. So they do not reflect on his integrity or independence, they just arose from his personality. He is an exceptionally cautious scientist. Thus, he would not accept that anything had any prospects of success unless entirely certain, or otherwise until tested and proved. This led him to say that it would be an invention to test a new vaccine platform for any target other than influenza or rabies (with their correlates of protection), not even RSV which he and his colleagues had selected in Geall A. et al, "Nonviral delivery of self-amplifying RNA vaccines" PNAS 2012; 109(36): 14604-14609 ("Geall 2012") and which was referenced in other publications as well. Likewise, he said it would be an invention to apply an established mRNA vaccine approach to any new target, and that anything going beyond the CGK would be inventive.
81. To be fair, some of this arose from his not being comfortable with what "obvious" specifically meant in the context of patents, and I can sympathise with that (and a similar point applied to Prof Dougan who got himself in a tangle over what "expect" connoted with or without the word "could"). But even allowing for it, it is plain that he was applying standards which were not the right ones for prospects of success and obviousness, and I cannot rely with confidence on his overall views on those matters. His evidence was nonetheless useful on some aspects of CGK and I found him a good explainer of the technology.
82. Dr Sola is the co-director of the Spanish National Centre for Biotechnology (CNB-CSIC) in Madrid. Her qualifications include a degree in biology, a Master's degree in bioengineering and a PhD in molecular virology. Since 2010 she has been Associated Professor, Master in Virology, at the Complutense University of Madrid. Between 2005 and 2013 she researched the molecular mechanisms of coronavirus transcription. From 2011 to 2022 she was co-principal investigator of a project entitled "SARS-CoV-host cell interactions and vaccine development" which looked at potential vaccine candidates based on live attenuated viruses for SARS-CoV and both live attenuated viruses and RNA replicons for MERS-CoV.
83. I found Dr Sola's demeanour during her oral evidence to be exemplary, but I accept Pfizer/BioNTech's first main criticism of her, which is that it is hard to understand how she offered the opinion that there was little interest in coronavirus vaccines at the EP565 Priority Date in the light of so many documents saying the opposite, and, materially to assessing her as a witness, including her own work (in particular a 2016 book chapter and comments at a symposium in 2014). This evident tension may be the explanation for some especially difficult answers, for example when she suggested at one point that if there was a need for MERS vaccines in 2013-2015 or so, it had disappeared in 2016, against the background that she had said in her first report that the CGK did not change between 2015 (the priority then claimed by Moderna for EP565) and 2016 (the filing date). Pfizer/BioNTech's other criticism of her was that she had the same inappropriately pessimistic outlook as Dr Ulmer. I agree with this. She equated a reasonable expectation of success with something near certainty and so she had the wrong standard in mind when offering her opinions.
84. Prof Dougan is an Emeritus Professor in the Department of Medicine at the University of Cambridge. He completed a biochemistry degree and PhD at the University of Sussex before undertaking postdoctoral research at the University of Washington and becoming a lecturer and researcher at Trinity College, Dublin. He worked for the Wellcome Foundation between 1983 and 1992, working on vaccines and antibodies for a number of diseases, and between 1992 and 2004 he was Professor of Physiological Biochemistry and Head of the Centre for Molecular Microbiology and Infection at Imperial College London. From 2004 to 2017 he was Head of Pathogens at the Wellcome Trust's Sanger Institute, where he identified numerous pathogenic antigens as vaccine candidates. In 2017 he became GSK Chair in Microbial Pathogenesis at the University of Cambridge. He has also been a member of, or chaired, several committees and advisory boards of companies and organisations, including WHO vaccine committees and the scientific board of Chiron. He has received awards for his work in the vaccine field and was elected as a Fellow of the Royal Society in 2012. Thus, Prof Dougan has had a career of extraordinary eminence but as a witness he had a number of serious shortcomings. Counsel for Moderna criticised him on a number of bases.
85. First, Moderna said that his demeanour was argumentative and hostile. I agree with this, but frustrating though it was, my task is to assess carefully how much it affected the substance of his evidence. In many ways it did not, other than to make the exercise more fractious and time consuming than it need have been. For example, his interruptions, (unjustified) accusations that the cross-examiner was seeking to mislead or trick him, and habit of answering questions with questions were not important to the objective exercise in hand. I do think that to some extent he was arguing a position, but on the key issues such as the CGK about vaccine targets and what was obvious from WO674 (leaving aside the hindsight point which I come to next), I think he was genuinely giving his own honest opinion.
86. Second, Moderna said that his selection of SARS and MERS from WO674 was driven by hindsight and that he could offer no proper explanation for it. I agree with this. I am talking now about pages 29-31 of WO674. When asked why he called out SARS from those pages he said "I do not know. I just did, yes." Then he said he had called out ones in the examples, but that does not explain SARS. At other points in his written and oral evidence he relied on similarity of viral glycoproteins and on commonality of respiratory diseases. None of this was at all convincing. I therefore conclude that Prof Dougan was bringing in hindsight in claiming that SARS and MERS stood out from those pages of WO674. However, this point is in a way something of a sideshow because of example 20 of WO674, which expressly calls out a MERS-CoV mRNA vaccine using the spike glycoprotein. This, coupled with my findings as to the CGK about the knowledge of SARS and MERS as serious threats of international concern, make the status of the list on pages 29-31 of low importance. The fact that Prof Dougan let hindsight in on this occasion without acknowledging it, and his unsatisfactory explanations mean that I have to be especially careful to scrutinise his other opinions for hindsight, and I have aimed to do so.
87. Third, Moderna said that Prof Dougan's expertise relevant to this dispute was limited, and in particular, that he had overplayed his knowledge of nucleic acid vaccine research gained on the Scientific Advisory Board of Chiron/Novartis. I do not think there was much, if anything, in this. I agree that the central thrust of Prof Dougan's work has been elsewhere over the years, but his experience and publications have been very wide ranging and while he was not giving evidence on his area of greatest speciality, I think he knew amply enough and had enough exposure to the science over the years, to be able to help the Court. On a related point, Moderna asked the forensic question: why could Pfizer/BioNTech not find and call a supportive witness who was closely and personally involved in nucleic acid vaccine research at the EP565 Priority Date? Why did they have to call someone with less familiarity in the person of Prof Dougan? Counsel for Pfizer/BioNTech had a convincing answer to this, though: anyone who matched that profile would all too probably have been working for a competitor, and moreover a competitor likely now to be in litigation with Pfizer/BioNTech.
88. There are many other individual points that Moderna made about Prof Dougan; these are only the main ones. Counsel for Pfizer/BioNTech encouraged me to look at "the substance and not the style" and indeed confined his submissions largely to the former. I do not think that will do. The problems of so-called "style" were significant and material and lead me to discount Prof Dougan's evidence to a real extent, and to exercise the particular caution about hindsight that I have indicated above. They do not however lead me to reject his evidence outright, or even nearly so. I do not know why he was so argumentative - perhaps it was nerves, or not being used to being challenged so searchingly or for an extended period - but while his objectivity was reduced, I do not think he was being at all dishonest and, importantly, I think he was a much better guide as to how a pragmatic vaccinologist who wanted to carry things forwards where there was appropriate scientific support would think and behave than was Dr Ulmer (and from the perspective of a virologist, Dr Sola) with their deep negativity. In addition, and as I have touched on already, I have an extensive documentary record of what was going on in the field to enable me to calibrate which of the side's experts' views are more reflective of how matters stood at the EP565 Filing Date. The documentary record clearly supports the position of Pfizer/BioNTech and Prof Dougan.
89. Professor Susan Weiss gave evidence from the perspective of a microbiologist/virologist with knowledge of coronaviruses.
90. Prof Weiss is a Professor of Microbiology at the University of Pennsylvania School of Medicine. She is also co-director of the Penn Center for Research on Coronaviruses and Other Emerging Pathogens.
91. Her PhD was on paramyxoviruses at Harvard and her post-doctoral research was on retroviruses at UCSF. She became an Assistant Professor at the University of Pennsylvania, where she is now, in 1980. She established a research program in coronaviruses and her laboratory has studied six of the seven known human coronaviruses, including MERS-CoV and SARS-CoV.
92. Professor Weiss's evidence went to a small part of the case. She was instructed to give the perspective of the coronavirus virologist whose role in the skilled team was to educate the team on betacoronaviruses in a way which would inform the choice of immunogen after the nucleic acid vaccinologist had already decided to develop a betacoronavirus nucleic acid vaccine.
93. She was cross-examined on topics which overlapped with Prof Dougan's evidence, such as the interest in and need for, betacoronavirus vaccines. Her experience is in relation to basic coronavirus research; she is not a vaccinologist.
94. Moderna sought to limit any reliance to be placed on Prof Weiss's evidence. Moderna submitted that Prof Weiss has not spent time on applied aspects of coronavirology, such as vaccine and antiviral work, and that she had very little knowledge or experience of immunology. These aspects were dealt with primarily by Prof Dougan.
95. During cross-examination, Prof Weiss stated that she agreed with Dr Sola in a general way that there was no interest in vaccine development for coronaviruses. Upon re-examination, she reverted to the position in her written evidence that there was interest in betaCoV vaccines and she was aware of colleagues who were interested in betaCoV vaccine development. Her position overall was plainly consistent with what she said in her written evidence and in her re-examination. I conclude that she must have misspoken or misunderstood in her cross-examination, given that the answer was an isolated statement and so far out of the flow of her clear general views. In passing I mention that the capitalisation of "betaCoV" in the papers for this case was not consistent and I have not tried to make it so in this judgment; sometimes it is "BetaCoV".
96. I found Prof Weiss to be a helpful witness. Her evidence was on a relatively minor part of the case but it was delivered in a clear and concise manner, and she was well qualified to give the evidence she did.
97. Professor Christopher Alabi gave evidence from the perspective of the skilled team's delivery chemist.
98. Prof Alabi is currently an Associate Professor at the Smith School of Chemical and Biomolecular Engineering at Cornell University. His work focusses on the assembly of macromolecules for use in drug delivery systems; this includes the use of lipid nanoparticles to deliver RNA. He studied chemistry and chemical engineering at undergraduate level before undertaking a PhD at CalTech relating to the targeted nanoparticle delivery of oligonucleotides to cancer cells.
99. Prof Alabi conducted post-doctoral research at MIT on lipid-based delivery systems for RNA and DNA. In 2013 he moved to Cornell University, where he is now, as an Assistant Professor.
100. I found Prof Alabi to be an excellent witness. He gave clear evidence and fairly accepted when issues fell outside of his expertise.
101. Moderna contended that all of Prof Alabi's work up to October 2016 related to siRNA and the purpose of siRNA is to stop the expression of the mRNA, which is in stark contrast to the reason for using mRNA in vaccines, where the aim is to produce the immunogen of the target protein. This was framed by Moderna as a minor caveat to Prof Alabi's suitability to give evidence on the encapsulation and delivery of RNA in LNPs. I found Prof Alabi to be highly knowledgeable and sufficiently capable of assisting the Court on the matters he was called upon to give evidence, therefore I reject this allegation.
102. It emerged in cross-examination of Pfizer/BioNTech's witnesses on EP565 that they had not seen each other's draft reports or met (remotely or in person) to discuss the makeup and approach of the skilled team. Instead, it seems that in some instances they were just told by the solicitors helping to prepare their evidence what each other would say. In the event I do not think this mattered to the substance of the case, although I bear it in mind, and Counsel for Moderna did not really make anything of it in oral closings. I will say, however, that this is not a satisfactory approach. On different facts, the interaction of the notional skilled team could be critical and it would be much better, if there are multiple experts, for them to conduct some sort of dialogue about this. A discussion should not be difficult to arrange at least remotely, but the bare minimum ought to be sight of advanced drafts of each other's reports so that each expert can consider and take account of the views of the other. This is still likely to be open to the criticism that it does not capture the substance of the real interactions that would take place, but it is certainly better than advisers merely relaying what another expert is planning to say, which is all too likely to be incomplete, subject to the perceptions of the advisers, and lacking in transparency.
103. The parties cited a number of authorities on the correct approach for determining the skilled person.
104. In Schlumberger v EMGS [2010] EWCA Civ 819 Jacob LJ considered whether it was right to have different skilled teams for obviousness and sufficiency. After reviewing a number of authorities, lastly the decision of Laddie J in Inhale Therapeutic Systems Inc v Quadrant Healthcare plc [2002] RPC 21, Jacob LJ concluded in paragraph 53 that:
What Laddie J. was saying was that where an invention involves the use of more than one skill, if it is obvious to a person skilled in the art of any one of those skills, then the invention is obvious. And rightly so, for it would otherwise impede a class of person who found it obvious. So here, if the invention was obvious to a CSEM expert alone or to a geophysicist alone, then the patent is invalid. Mr Thorley did not contend otherwise. What is important to note is that Laddie J. was careful to recognise that there could be invention in marrying together concepts from unrelated arts. A non-obvious marriage of skills is essentially what Mr Thorley is contending for here.
105. Thus Jacob LJ confirmed that there can be circumstances where the skilled person can be different for the purposes of obviousness and sufficiency. But ultimately, neither party at this trial contended that the skilled person should be defined differently for the purposes of assessing obviousness and sufficiency.
106. In Novartis v MedImmune [2012] EWCA Civ 1234, in the Court of Appeal Kitchin LJ (as he then was) considered the right approach regarding the skilled team at [73] to [77]:
73. As the judge explained, in this case there was a dispute as to the identity of the team to whom the patent is addressed. MedImmune contended it is addressed to a team consisting of an immunologist and a molecular biologist, perhaps assisted by a chemist. Novartis argued the patent is addressed to a team of scientists with differing backgrounds in areas such as immunology, in particular antibody structural biology, molecular biology and protein chemistry, but with a common interest in antibody engineering. As the judge identified, the essential difference between the two formulations lies in the degree of specialisation of the team in the field of antibody engineering.
74. The judge preferred Novartis' submission on the basis that the evidence showed that real research teams in the field were teams of the kind contended for by Novartis. He added that, in his view, the specification of the patent is consistent with this characterisation of the skilled team.
75. MedImmune contended that the judge fell into error in so finding because the invention has a broad application and is not confined to antibody engineering. It continued that expertise in immunology and molecular biology is sufficient to implement its teaching.
76. I have no doubt that the judge identified the skilled team correctly. As Jacob L.J. explained in Schlumberger Holdings Ltd v Electromagnetic Geoservices AS [2010] EWCA Civ 819, [2010] RPC 33 at [42], the court will have regard to the reality of the position at the time and the combined skills of real research teams in the art. A little later, at [53], he continued that where the invention involves the use of more than one skill, if it is obvious to a person skilled in the art of any one of those skills, then the invention is obvious. Finally, at [65], he explained that in the case of obviousness in view of the state of the art, a key question is generally "what problem was the patentee trying to solve?" That leads one in turn to consider the art in which the problem in fact lay. It is the notional team in that art which is the relevant team making up the person skilled in the art.
77. The judge found that by 1990 antibody engineering was an established field. The three leading teams were those led by Dr Winter at the MRC Laboratory of Molecular Biology and CAT, by Professor Lerner at the Scripps Institute and by Andreas Plückthun at the Max-Planck-Institut für Biochemie. Other teams were also interested, including the research group led by Professor Stefan Dübel at the Deutsches Krebsforschungszentrum and teams at Genentech, Genex Corporation, Ingene, SmithKline Beecham and Genetics Institute. All of these teams were likely to have a practical interest in the subject matter of the invention, in methods for preparing binding molecules including, specifically, antibodies and fragments of them, and selecting those with specificity for particular antigens. They had a need for a system which would allow them to screen very large numbers of different binding molecules. The invention was therefore plainly of interest to antibody engineers and the fact that it may have a broader application is neither here nor there.
107. In its written opening, Moderna relied on the fact that MedImmune's attack on the Judge's reasoning failed, relying in particular on Kitchin LJ's reasoning in [77] on this.
108. Moderna also relied on Actavis v Lilly [2015] EWCA Civ 555 in their opening skeleton as showing that in an appropriate case, different claims might engage different skilled persons. Floyd LJ set out the parties' positions at [31]:
[31] Lilly contends that the 508 patent is addressed to an oncologist, whereas Actavis contends that it is addressed to a team which, although it includes an oncologist, also includes a chemist. The judge preferred Actavis' argument on this point.
109. At [33] and [34] he went on to say:
33. I have no doubt that the judge was right to find that the patent is addressed to a team which includes a chemist in addition to the oncologist. Firstly, the invention requires the use of the pemetrexed disodium, and the specification of the 508 patent expects the skilled person to be able to obtain it and make it into an injectable solution. It is no answer to say, as Lilly does, that the claim is only framed in relation to manufacture because of a legal fiction concerned with the restriction on patentability of second medical use inventions. However the claim is formulated, the skilled addressee needs to manufacture the medicament and the manufacturing step is an essential requirement of the claim, necessary to prevent the claim falling foul of the method of treatment exclusion from patentability. In the absence of evidence that pemetrexed disodium was generally available, manufacture of the medicament includes making the active ingredient. Secondly, as the judge found, the teams who deal with developing and making medicaments for use in treatment in the real world comprise specialists in a range of disciplines, and in this context would comprise both a medical oncologist and a chemist. Finally, there is a fundamental inconsistency as the judge pointed out, arising out of Lilly's case that the claim would be understood as extending to the use of active ingredients other than pemetrexed disodium. Assuming for a moment that Lilly is correct, the evidence showed that choice of an appropriate alternative salt would not be something that the medical oncologist could assist with.
34. I would prefer not to express any concluded view on whether any weight can be attached to Actavis' argument based on claim 12, which attracted the judge. If the only reason for engaging the discipline in question were the existence of a separate set of claims, or a subsidiary claim, I would be disinclined to hold that the same addressee was necessarily required for all claims. But as I am of the view that claim 1 requires a chemist in any event, I need not explore that question further.
110. It was noted by Counsel for Pfizer/BioNTech that Floyd LJ did not in fact have to decide whether the formulation claim in claim 12 would have made any difference. Counsel for Pfizer/BioNTech also pointed out that in a later decision, Conversant v Apple [2019] EWHC 3266 (Pat), Birss J said at [32] that it was unusual but not wrong in principle for the skilled team to change as a result of a claim amendment (in that case limiting to smart phones and so excluding PDAs). I do not think I need to, or should, make any definite decision on the law on this point of detail, because given the claims in issue by the end of the trial it does not make any difference. It has no impact on EP949 and on EP565 the only claims that matter are claims 1 and 10, where the former is to a vaccine and the latter adds the requirement that the vaccine has a protective effect. This difference cannot affect the members of the skilled team for EP565.
111. Both parties relied on Illumina v MGI [2021] EWHC 57 (Pat). Counsel for Pfizer/BioNTech relied on [62] where Birss J (as he then was) explained why framing the art in a narrow way can go wrong by reference to Folding Attic Stairs [2009] EWHC 1221 (Pat) and the deputy Judge's example of the two-hole blue Venezuelan razor blades, and on [63] which states:
63. So while Folding Attic Stairs neatly explains one of the difficulties, given its facts the judge did not have to identify a principle to be applied to solve it. Furthermore, while a too narrow definition could be unfair to the inventors, it could be just as wrong and unfair to the public to define a team so widely that their common general knowledge is so dilute as to make something seem less obvious than it really was (see Pumfrey J. in Mayne Pharma Ltd v Debiopharm SA [2006] EWHC 1123 (Pat) at [3]-[4]).
112. Birss J went on at [66] and [67]:
66. In the present case Illumina proposed, based on Medimmune, that a sensible test was to require something which could properly be called an established field at the priority date. Depending on the facts the field could be a research field as in Medimmune or a field of manufacture as in Folding Attic Stairs.
67. The advantage of this test is that it provides a principled way of solving the problem identified in Folding Attic Stairs. If the design and manufacture of folding attic stairs in particular was an established field then there is nothing unfair in defining the skilled person that way. But if not then the wider definition (general carpenter plus metal fabricator) is appropriate. In other words the width of the field in which the skilled person operates for the purposes of obviousness (a.k.a. the "art in which the problem lay" (per Schlumberger)) is ultimately governed by what was actually going on up to the priority date. It is not primarily a function of the invention itself, the problem to be solved, nor the patent's text.
113. It was common ground between the parties that the proper approach to take when defining the skilled person for the purposes of obviousness is set out by Birss J in Illumina at [68]:
68. I conclude that in a case in which it is necessary to define the skilled person for the purposes of obviousness in a different way from the skilled person to whom the patent is addressed, the approach to take, bringing Schlumberger and Medimmune together, is:
i) To start by asking what problem does the invention aim to solve?
ii) That leads one in turn to consider what the established field which existed was, in which the problem in fact can be located.
iii) It is the notional person or team in that established field which is the relevant team making up the person skilled in the art.
114. Both parties agreed that the principles identified above also apply when the skilled person is the same for obviousness and sufficiency.
115. Finally, both parties relied on Alcon v Actavis [2021] EWHC 1026 (Pat). Pfizer/BioNTech referred me to paragraph 23 of Moderna's opening skeleton with which they agreed and which read:
23. In Alcon v Actavis [2021] EWHC 1026 (Pat), this Court approved and applied Birss J.'s summary of the applicable principles in Illumina and emphasised at [31] the following four points (paraphrased and with citations added):
a) there are requirements:
i) not to be unfair to the patentee by allowing an artificially narrow definition: Illumina at [62] and Folding Attic Stairs v The Loft Stairs Company Ltd [2009] EWHC 1221 (Pat), [2009] FSR 24 at [33]-[34], and
ii) not to be unfair to the public (and the defendant) by going so broad as to "dilute" the CGK (Mayne Pharma Ltd v Debiopharm [2006] EWHC 1123 (Pat) at [4]).
There is therefore an element of value judgment in the assessment.
b) The Court must consider the real situation at the priority date, and in particular what teams existed: Schlumberger at [42].
c) The need to look for an "established field", which might be a research field or a field of manufacture: Illumina at [65]-[66] and Medimmune v Novartis [2012] EWCA Civ 1234, [2013] RPC 27 at [73]-[76].
d) The starting point is the identification of the problem that the invention aims to solve: Schlumberger at [65], Illumina at [60]-[61].
116. Pfizer/BioNTech, however, did not agree with Moderna's conclusion in paragraph 24 that "The skilled team will be defined by the narrowest sub-set that actually comprised an 'established field' at the time, and no wider". I agree with Pfizer/BioNTech that such a rigid rule would be inconsistent with the principle that a value judgment is involved, as explained above, but in general if there is clear factual evidence of an established field that embraces the problem to be solved, that is bound to be an important factor and will often hold sway.
117. As will be clear from my treatment of the witnesses, above, in relation to EP949 the parties called one expert each, albeit Pfizer/BioNTech made some arguments that Prof Rosenecker was the "wrong" member of a/the team, whereas on EP565 the parties called multiple experts and it is clear there would be a team in real life. I have not tried to achieve perfect consistency in the use of "person" or "team" in this judgment, conscious that both are abstractions and what ultimately matters is the overall CGK, skills and attitudes, but I have borne in mind that on EP565 in particular a team would be involved and so I have addressed the individual roles below.
118. For reasons explained above in the section of this judgment on case management, I asked each side to put in a single set of closing submissions on the law of added matter and of novelty, to seek to ensure consistent treatment in relation to EP949 and EP565.
119. There was a good deal of agreement, as one would hope.
120. It was agreed that the fundamental approach to added matter is as set out in Nokia v IPCom [2012] EWCA Civ 567. Kitchin LJ encapsulated the overall test at [60]:
Ultimately the key question is once again whether the amendment presents the skilled person with new information about the invention which is not directly and unambiguously apparent from the original disclosure. If it does then the amendment is not permissible.
121. That is consistent with other earlier statements such as Richardson-Vicks' Patent [1995] RPC 568 at 576, but I do not think it is necessary to go back to earlier formulations and Kitchin LJ's has the advantage that it explicitly calls out the standard (directly and unambiguously) that has to be met. A structured approach to addressing this question is to be found in Bonzel v Intervention [1991] RPC 553. It is frequently used but, just as with the Pozzoli approach to obviousness, it is not mandatory; neither side used it at this trial but nothing turns on that.
122. It was also agreed that the EPO approaches added matter using the "gold standard". A convenient statement of this can be found in the Case Law of the Boards of Appeal of the EPO (10th Ed) at 1.3.1:
1.3.1 Gold standard: directly and unambiguously derivable
Any amendment to the parts of a European patent application or of a European patent relating to the disclosure (the description, claims and drawings) is subject to the mandatory prohibition on extension laid down in Art. 123(2) EPC and can therefore, irrespective of the context of the amendment made, only be made within the limits of what a skilled person would derive directly and unambiguously, using common general knowledge, and seen objectively and relative to the date of filing, from the whole of these documents as filed (G 3/89, OJ 1993, 117; G 11/91, OJ 1993, 125; G 2/10, OJ 2012, 376, referring to this test as "gold standard"; confirmed for disclosed disclaimers in G 1/16, OJ 2018, A70; for undisclosed disclaimers, see however chapter II.E.1.7.2 c)). After the amendment the skilled person may not be presented with new technical information (G 2/10).
123. The parties agreed that the disclosure test for added matter is in substance, for present purposes at least, the same as that for anticipation (this is subject to the qualification, which does not affect the arguments or my analysis, that for anticipation it is enough that the prior art discloses something within the claim but to avoid added matter it is not enough that the prior application discloses some of the relevant subject matter). From a UK perspective in relation to novelty, the "clear and unmistakable" standard dates back many years, the two most frequently cited decisions being General Tire & Rubber Company v Firestone Tyre & Rubber Company Ltd [1972] RPC 457 and Synthon v SmithKline Beecham [2005] UKHL 59, [2006] RPC 10.
124. In fact I should mention, as pointed out by Moderna, that there has over the years been a complex discussion in the EPO case law about whether at a very subtle level the added matter standard is identical to or different from the novelty standard. At one time it was said that one could apply a "novelty test" for added matter but that has faded with the clear application of the gold standard and statements at the highest level of authority from the EPO that other tests may sometimes be appropriate but do not displace the gold standard.
125. In some of its earlier submissions, Moderna referred to earlier EPO cases which used a "serious contemplation" test. In those submissions Moderna both relied on the test (for EP565) and deprecated it (for EP949), but as a result of the parties' single sets of submissions Moderna disclaimed any reliance on it, and Pfizer/BioNTech did not rely on it either. It was the subject of some discussion in UK case law, in particular in Jushi Group v OCV [2018] EWCA Civ 1416 where Floyd LJ said it was unobjectionable as long as it did not "relax the rigour of the general law of novelty". Although it has no impact on my analysis given that none of the parties at this trial relied on it in the end, I think the "serious contemplation" formulation is best avoided: it does not add anything if applied correctly, but its phrasing makes it sound like a different test from the clear and unambiguous standard.
126. The gold standard formulation above refers to the whole document and to the common general knowledge. I accept, of course, that the whole document has to be considered, but that does not mean that it is a reservoir from any part of which a feature can be taken to combine with a feature from some other part, in the absence of a clear teaching to do so. Similarly, the CGK informs, as ever, what the skilled person understands from the document but it does not make the CGK a reservoir from which features can freely be drawn to be plugged in at will. I return to the role of CGK in a little more detail in relation to selection from multiple lists at paragraphs 141 and 142.
127. Prior documents (the prior art for novelty, the application as filed for added matter) may contain disclosures in the form of lists, or groups. The way in which these situations are to be addressed has been considered by the UK courts and by the EPO.
128. In their submissions on the applicable law both sides referred on this point to the decision of the Court of Appeal in Dr Reddy's v Eli Lilly & Co [2010] RPC 9. It is helpful because it considers the EPO position and it is useful to quote from it quite extensively:
23. Olanzapine is one of the 1019 compounds of formula (I) and one of the 86,000 compounds of the "preferred" class. It is not mentioned specifically.
24. DRL contends that nonetheless this specific compound lacks novelty - that in the language of EPC Art.54 it formed "part of the state of the art" having been "made available to the public by means of a written ... description." The contention amounts to this: that every chemical class disclosure discloses each and every member of the class. It would, it seems, even apply if the formula had simply been written down without any suggested utility.
25. I reject the contention for two reasons: firstly as a matter of a priori reasoning and secondly because it is inconsistent with settled EPO Board of Appeal case law.
26. First then, the a priori considerations apart from case-law. An old question and answer runs as follows: "Where does a wise man hide a leaf? In a forest." It is, at least faintly, ridiculous to say that a particular leaf has been made available to you by telling you that it is in Sherwood Forest. Once identified, you can of course see it. But if not identified you know only the generality: that Sherwood Forest has millions of leaves.
27. The contention has no logical stopping place. If there is disclosure of olanzapine here, why would one not regard an even more general disclosure as a disclosure of it. Suppose the prior art had merely been of "3-ringed organic compounds?" Such a description would encompass much much bigger numbers than the 1019 of formula I. Yet the logic of the argument would be the same - that there is a disclosure of each and every member of the class.
28. I would add that I would regard the listing out of a great number of compounds as opposed to the use of a Markush formula in the same way. To say a particular book is identified by saying "the books in the Bodleian" is no different from saying it is identified by providing access to the catalogue of the Bodleian.
29. Similarly it makes no sense to say that a generalised prior description discloses a specific matter falling within in. The judge's example illustrates the point. A prior disclosure of "fixing means" is not a disclosure of a particular fixing means e.g. welding or riveting even though you could list out a whole number of ways of fixing things together which would include these means.
30. Thus logic dictates rejection of the argument that a disclosure of a large class is a disclosure of each and every member of it. So also does EPO case-law. Mr Carr accepted that was so, so I can take the matter quite shortly, going to just one case, Hoechst/Enantiomers T 0296/87, 30 August 1988 , which effectively sums up earlier cases. It said:
"6.1 Here the Board is guided by the conclusions it reached in its 'Spiro compounds' decision T 181/82 (OJ EPO 1984, 401) concerning the novelty of chemical entities within a group of substances of known formula. With regard to products of the reaction of specific spiro compounds with a (C1-C4)-alkyl bromide defined as a group, the Board drew a sharp distinction between the purely intellectual content of an item of information and the material disclosed in the sense of a specific teaching with regard to technical action. Only a technical teaching of this kind can be prejudicial to novelty. If any such teaching is to apply in the case of a chemical substance, an individualised description is needed."
So what one must look for by way of an anticipation is an "individualised description" of the later claimed compound or class of compounds. This case is miles from that. It is noteworthy that the Board's application of that principle in that case to enantiomers was specifically followed by this Court in Generics (UK) Ltd v H. Lundbeck A/S [2008] EWCA Civ 311; [2008] RPC 19 per Lord Hoffmann at [9].
31. It is not necessary here to go into what is sufficient to amount to an "individualised description." Obviously the question may partly be one of degree, but other considerations may come in too, for instance the specificity of any indicated purpose for making the compounds. A mere woolly indication of the possible use of the prior class may require less specificity than a precise one.
32. This view of the law accords with the decision of the House of Lords in SmithKline Beecham plc's (Paroxetine Methanesulfonate) Patent [2006] RPC 10 Lord Hoffmann said:
"[22] If I may summarise the effect of these two well-known statements, the matter relied upon as prior art must disclose subject-matter which, if performed, would necessarily result in an infringement of the patent. That may be because the prior art discloses the same invention. In that case there will be no question that performance of the earlier invention would infringe and usually it will be apparent to someone who is aware of both the prior art and the patent that it will do so."
Where you have a patent for a particular chemical compound and a prior art general disclosure, performance of the general disclosure (which means no more than using anything within it) does not necessarily result in infringement of the patent. In this case, for instance, you can "perform" 235 in any of 1019 ways - only one of them would result in infringement of the later patent.
33. Accordingly I would reject the anticipation attack. In so doing I am glad to find that the approach I adopt is not only the same as that in the EPO but also the same as that in Germany. It was well-articulated in the case involving the German equivalent of the patent in suit, STADApharm No.1-2W 47/07 (29 May 2008). The Bundespatentgericht had held the patent invalid. But even so the Oberlandsgericht in Dusseldorf (an infringement only court) decided to enforce the patent, holding in effect that the Bundespatentgericht's decision was so obviously wrong that it would surely be reversed on appeal (which it duly was). In the context of novelty over 235 it said:
"what is relevant is whether by the indications given by a prior art document on the chemical compound, the skilled person is able without difficulties to carry out the invention relating to this compound, i.e. whether he can actually obtain the relevant substance. For merely practical reasons the availability cannot be confirmed by stating that the person skilled in the art was able to work through the countless alternatives falling under this formula, and in doing so would eventually arrive at the compound in question. It is disclosed to him in a cited reference only when the prior document contains a concrete indication of the claimed compound, such as a description thereof as a preferred embodiment, and if the skilled person is able to produce the compound on the basis of this indication and his general technical knowledge ."
On the appeal the Bundesgerichthof did not even find the allegations of invalidity over 235 as worthy of mention - perhaps they had been dropped by then.
129. I found particularly helpful and relevant to the overall test ("individualised disclosure"), the idea of the contrast between "purely intellectual content" and "specific teaching" (from Hoechst T296/87), the guidance that the question is one of degree, and the pointer in [31] that the specificity of any indicated purpose can be relevant. I also note that the Court of Appeal was clear that there is no conceptual difference between a list and the identification of a group such as a Markush formula (at [28]).
130. These principles have been applied on numerous occasions in the UK and the EPO. The sheer number of members of the list or class where there has or has not been anticipation has varied widely. There were 1019 possibilities in Dr Reddy's and that is an easy and obvious case for saying no individualised disclosure, but at the other end of the spectrum the disclosure of a racemate in Hoechst T296/87 was held not to be a disclosure of an enantiomer. A case in the middle is Almirall v Boehringer [2009] FSR 12 where 159 examples were held each to be sufficiently individualised (although the novelty attack failed because it was necessary to combine the right member with other features).
131. Each side before me accepted that there is no fixed numerical cut-off for individualisation (although this did not stop them bandying about small, large and middling numbers from cases). I will proceed on the basis that the overall test is whether there is an individualised disclosure and that the size of the list/class is one relevant factor. Often, no doubt, it will be a major factor and in the right case it might be decisive.
132. A facet of individualised disclosure which also arises quite often concerns selection from multiple lists. For example, a Markush formula may provide a list of options for position R1 and another list for position R2, with the inquiry (whether for the purposes of added matter or novelty) being whether a compound with a particular choice at R1 and another particular choice at R2 is clearly and unambiguously disclosed.
133. Moderna referred me to the early decision in T12/81 to the following effect (reason 130):
...If on the other hand two classes of starting substances are required to prepare the end products and examples of individual entities in each class are given in two lists of some length, then a substance resulting from the reaction of a specific pair from the two lists can nevertheless be regarded for patent purposes as a selection and hence as new.
134. Moderna also helpfully provided references showing that the approach is applied by the EPO both to novelty and added matter, although this was not in dispute.
135. In the UK, the issues presented by multiple lists have been recognised in a number of cases, including in particular GSK v Wyeth [2016] EWHC 1045 (Ch [sic]) and MSD v Shionogi [2016] EWHC 2989 (Pat). In the former, Henry Carr J said this at [119]:
In my judgment, selections from two or more lists may well amount to impermissible added matter, but this is not a rigid rule. In order to see whether there is a new combination of independent features from two or more lists, the whole contents of the application as filed must be considered, including its general disclosure. It is necessary to avoid a mechanistic approach, and to compare the disclosures of the application as filed and the patent, through the eyes of the skilled person, in order to answer the overall question of whether the skilled person would learn new technical subject matter which was not disclosed in the application.
136. The important points from this are that the approach is not mechanistic and that the underlying question remains whether there is new information disclosed (the context there being added matter). This is no different from the EPO, however, which also recognises that the approach is not formalistic (in the sense that a choice from two lists does not automatically mean there is not individualised disclosure) and that the ultimate question is still clear and unambiguous disclosure. See T 783/09 at [5.6]:
However, given the term "can" in the citation from decision T 12/81, the absence of a direct and unambiguous disclosure for individualised subject-matter is not a mandatory consequence of its presentation as elements of lists. Thus, the "disclosure status" of subject-matter individualised from lists has to be determined according to the circumstances of each specific case by ultimately answering the question whether or not the skilled person would clearly and unambiguously derive the subject-matter at issue from the document as a whole
137. I do not think any of these basic aspects about selecting from lists was in dispute; the parties cited a number of different EPO cases but both cited the above passage from T783/09.
138. One way in which application of the two lists principle might be unduly mechanistic and out of keeping with the underlying gold standard question would be if all possible combinations arising from the twin choices were artificially treated as being of equal significance when the disclosure of the document in question placed more emphasis on some than others. Thus the EPO case law recognises that there may be implicit or explicit "pointers". Various such pointers have been recognised by the EPO, and each side before me referred to the Case Law at II.E.1.6.
139. I do not think there is any conceptual limit on what may be a pointer in this sense, but a particularly common one is a statement of preference within a list in the document in question. I note however that in a number of the EPO cases it was held that there was added matter in combining a preferred member of one list with a member of another list for which no preference was expressed. In general, too, what the EPO looks for is a pointer to the combination; this cannot be an absolute rule, but it makes sense. See for example the references to T2273/10 and T1032/12 on pages 524 and 525 of the Case Law.
140. Other pointers could potentially be dependent claims (see e.g. T583/93), or members of a list which feature strongly in the preferred embodiments (in T583/93 the Board linked these by saying that dependent claims are inherently indicators of preferred embodiments - see 4.7 of the reasons), but again there cannot be a rigid rule. I do not think at this level that there was any disagreement between the parties at this trial.
141. In relation to pointers, Moderna argued that it was not legitimate to say that there was a relevant pointer merely because the CGK would be that one among a number of choices was desirable, see e.g. Euro-Celtique T3035/19 at 1.73:
The argument of the appellants amounts to inferring from common general knowledge a pointer to this particular selection in the absence of any justification therefor in the earlier application as filed. In this the Board shares the position of the respondents that a reference to the common general knowledge cannot compensate for the lack of disclosure in the application itself.
142. Pfizer/BioNTech accepted this and I agree. This is the difference between using the CGK to assess the skilled person's understanding of what is disclosed, and using the CGK as a reservoir of additional disclosure. The former is mandatory and the latter is illegitimate.
143. As with the other matters discussed above, "pointers" are a facet of the gold standard test, and not a replacement of it (see e.g. T1476/15 at 3.6).
144. The EPO case law concerning selection from multiple lists sometimes, including in T12/81, refers to the lists being "of some length", but as with individualised disclosure generally there cannot be any particular numerical requirement for this, and there are instances where the lists have been short yet a specific combination has been held not to be disclosed (e.g. T686/99 Lubricant for Refrigerating Oils, one list of three oil types and two refrigerants, T7/86 Xanthines, 2x5).
145. It is also important to have in mind whether two lists are independent or not; it may be that the choice from one list affects the choice from another. I accept this as a general proposition, but need to comment on one particular case relied on by Pfizer/BioNTech, which was T1581/12. There, the issue was added matter (under Article 76 EPC, relating to divisional status) and the question was whether it amounted to a selection from two independent lists to focus in on one amino acid sequence from among a number, and "combine" it with a particular fragment length. The opponent said this was combining two lists (one of the sequences, one of lengths) but the Board disagreed: it said that the disclosure of the full sequences inherently disclosed all fragments thereof so no recourse to a list of lengths was needed. I agree that this is an instance of two lists not being independent, on the Board's analysis of the facts. At first blush, the notion that the full length sequences implicitly disclosed all fragments looks generous to the patentee, but I do not have a full picture of the facts there and so assume that it is correct. I can see that on that basis there was reason to say that it was not a case of combining two lists, but all I derive from the case is that if on examination there is no need to combine two lists, then the principle does not apply. Pfizer/BioNTech tried to use this case for a different purpose, which was to say that in relation to novelty of EP949 over UPenn, the disclosure of use of a modified nucleotide necessarily disclosed all percentage replacements of the natural nucleotide, from 0% to 100%. This is a false analogy with T1581/12 and an illegitimate attempt to elevate the specific facts of that case to a point of principle. It is a false analogy because while the sequence ABCDEFG might be said also to disclose the sequences ABCDE and ABCDEF within it and inherently with them their lengths, the mere disclosure of using a modified nucleotide does not necessarily say anything at all about the percentage replacement, it just gives the identity of the modification, if it is made. I agree that given the instruction to use a modified nucleotide the skilled person would themselves have to choose a percentage if they made the modification, but that is nothing to do with unambiguous disclosure of the document.
146. Finally, and at the risk of repetition, the test of clear and unambiguous disclosure is emphatically not an obviousness test. Pointers, in particular, are a facet of deciding the question of clear and unambiguous disclosure and not a licence for holding something to be disclosed merely because it was an obvious choice. This may be easier to say than to apply, but on novelty of EP949 I think it is of some importance.
147. There was no material dispute between the parties with regards to the basic approach to obviousness as set out in the decision of the Supreme Court in Actavis v ICOS [2019] UKSC 15 at [52] - [73], with its endorsement at [63] of the statement of Kitchin J, as he then was, in Generics v Lundbeck [2007] EWHC 1040 (Pat) at [72]:
The question of obviousness must be considered on the facts of each case. The court must consider the weight to be attached to any particular factor in the light of all the relevant circumstances. These may include such matters as the motive to find a solution to the problem the patent addresses, the number and extent of the possible avenues of research, the effort involved in pursuing them and the expectation of success.
148. At [63] to [73] of Actavis v ICOS, Lord Hodge expanded on Kitchin J's list of factors. Moderna summarised Lord Hodge's nine points in its closing skeleton for EP949 as follows:
i) whether something was "obvious to try": [65];
ii) the routine nature and/or established practice/s of research: [66];
iii) the burden and cost of the research programme: [67];
iv) the necessity for and the nature of the value judgments the skilled team would have to make in the course of a testing programme: [68];
v) the existence of multiple or alternative paths of research: [69];
vi) the skilled person's motive: [70];
vii) whether the results of research are surprising or unexpected: [71];
viii) avoidance of hindsight, particularly if the analysis is approached step-by-step: [72]; and
ix) whether a feature is a bonus effect or added benefit where the claimed invention is otherwise obvious: [73].
149. Moderna focussed particularly on the skilled person's motive and expectation of success in their closing skeleton for EP949. With regards to the skilled person's motive, it pointed to Hickman v Andrews/WORKMATE [1983] RPC 147 where Goff LJ for a unanimous Court of Appeal said at 189:
There is another preliminary question and that is what the expert is supposed to be doing. It cannot be that he is to look to the whole store of his imaginary knowledge and see if it is obvious to turn something therein to better account. He must think I have some definite object in view, and in the instant case, for example, he would be seeking to make a workbench which would be an improvement on those already on the market.
150. Moderna also quoted Sir Donald Nicholls VC in Mölnlycke v P&G [1994] RPC 49 at 114:
...obviousness connotes something which would at once occur to a person skilled in the art who was desirous of accomplishing the end.
151. In Moderna's EP565 closing it also referred to Hoechst Celanese Corp v BP Chemicals [1997] FSR 547 where Laddie J reviewed several authorities considering motive and concluded at 572:
All of those passages are consistent with the Object/Solution approach to obviousness adopted by the Technical Board of Appeal of the EPO. Even if the step from the prior art is a small one, to prove obviousness it is necessary to demonstrate that there is some reason for taking it.
152. I agree that these cases identify the principles that in general the skilled person looks for a practical result and requires a motive to do something. This is not an invariable rule, however: cases of AgrEvo obviousness and workshop modifications (though not relevant to the present case) are exceptions where achieving a result and motivation do not really enter the picture, or only to a very limited extent.
153. When emphasising Lord Hodge's eighth point, hindsight, Moderna referred to Gedeon Richter plc v Bayer Schering Pharma AG [2011] EWHC 583 (Pat), where Floyd J stated at [114]:
I think that the guiding principle must be that one has to look at each putative step which the skilled person is required to take and decide whether it was obvious. Even then one has to step back and ask an overall question as to whether the step by step analysis, performed after the event, may not in fact prove to be unrealistic or driven by hindsight.
154. At [69] of Actavis v ICOS Lord Hodge stated that "the existence of alternative or multiple paths of research will often be an indicator that the invention contained in the claim or claims was not obvious." He warned that "it is necessary to bear in mind the possibility that more than one avenue of research may be obvious" and endorsed the statement of Laddie J in Brugger v Medic-Aid Ltd (No 2) [1996] RPC 635 at 661:
[I]f a particular route is an obvious one to take or try, it is not rendered any less obvious from a technical point of view merely because there are a number, and perhaps a large number, of other obvious routes as well.
155. This statement does not mean that the existence of a number of ways forward is irrelevant. Kitchin J explicitly called out the "number and extent of the possible avenues of research" as a relevant factor in Generics, as quoted above. Whilst Laddie J's statement in Brugger should be kept in mind, it must not be taken too far.
156. Pfizer/BioNTech relied on the principle that there can be no invention in doing what is suggested in the prior art unless there is an established prejudice against that idea - the so-called "lion in the path" (see e.g. Pozzoli v BDMO [2007] EWCA Civ 588 at [24]-[29]). The principle is an important one, but it applies once a specific suggestion has been identified. It does not mean that there cannot be invention in, at a prior stage, choosing one among a large number of possibilities. So the point runs much better against EP565 where the question is largely whether it was obvious to do Example 20 in WO674, than against EP949 where a choice has to be made among numerous different modified nucleotides in the prior art.
157. I was referred in Pfizer/BioNTech's opening skeleton on EP949 to the structured approach of the Court of Appeal in Pozzoli. It requires, for each piece of prior art, that the tribunal proceed in the following way.
(a) Identify the notional "person skilled in the art";
(b) Identify the relevant common general knowledge of that person;
(c) Identify the inventive concept of the claim in question or if that cannot readily be done, construe it;
(d) Identify what, if any, differences exist between the matter cited as forming part of the 'state of the art' and the inventive concept of the claim or the claim as construed; and
(e) Viewed without any knowledge of the alleged invention as claimed, do those differences constitute steps which would have been obvious to the person skilled in the art or do they require any degree of invention?
158. In general, however, the parties did not thereafter structure their main submissions around the Pozzoli approach. It is not mandatory and I do not think it caused problems that the parties proceeded otherwise (for example by using the factors identified in Actavis v ICOS as a checklist). There are reasons to do with the nature of the prior art attacks that mean they are not the best fit with the Pozzoli approach in this case, for example in EP565 the issue is not so much differences at the level of claim features as whether the skilled team would do what the prior art says at all, and on EP949 there are some differences (percentage substitution, polyA tail) which would be identified by the Pozzoli approach but which are not in fact in play because they were accepted to be obvious if the key decisions about looking for and identifying other modified nucleotides and which to choose were made.
159. Where appropriate I have mentioned aspects of the Pozzoli approach below, but I have not tried to shape my analysis on obviousness as a whole to it given that the parties did not.
160. In keeping with current practice in the Patents Court, there was a joint document which identified the CGK that was agreed (the "ASCGK") and another identifying what was in dispute.
161. There was no general dispute about the law applicable to CGK. To be CGK, something must be generally known and accepted as a good basis for further action.
162. Many organisms store hereditary information in the form of DNA (deoxyribonucleic acid). DNA is a polymer comprised of two polynucleotide chains which coil round each other in the form of a double helix. Each chain is made up of monomeric units called nucleotides. The nucleotides in turn contain nucleobases, each attached to a five-carbon sugar (deoxyribose) and a phosphate group.
163. The four types of nucleobase in DNA are: adenine (A), thymine (T), cytosine (C) and guanine (G). A nucleobase combined with the sugar, but not the phosphate, is called a nucleoside. In summary:
Nucleobase = A, T, C or G
Nucleoside = nucleobase + five-carbon sugar
Nucleotide = nucleoside + mono, di or tri-phosphate
Thus the difference between nucleoside and nucleotide is the phosphate(s). For most purposes of the arguments in this case that is not material and so the same point could be made by referring to "modified nucleoside" or "modified nucleotide", and the usage was not entirely consistent, but it is unimportant.
164. The structures of the nucleobases are shown below. The bases are nitrogen-containing ring compounds, either pyrimidines (left) or purines (right). The pyrimidine uracil is not found in DNA but is found in place of thymine in RNA. Nucleotides with structures comprising these classic bases are known as "canonical" nucleotides, to differentiate them from naturally occurring nucleotides that have undergone post-transcriptional modification, discussed below, which are called "non-canonical".
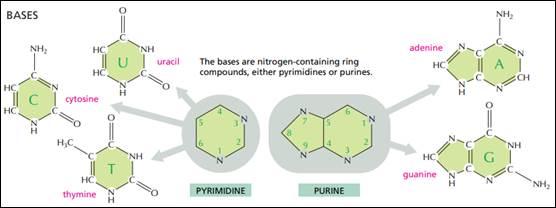
Structures of the nucleobases.
165. A nucleotide is illustrated below (in this case deoxyadenosine monophosphate) with the carbon atoms of the sugar moiety labelled in red according to standard notation:
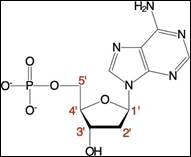
Structure of deoxyadenosine monophosphate.
166. In DNA, nucleotides containing these four bases are strung together in each polymer chain. The nucleosides are linked via phosphate bonds, thereby creating a backbone made up of alternating phosphate groups and deoxyribose units with a 5' phosphate at one end and a 3' hydroxyl at the other end as illustrated below:
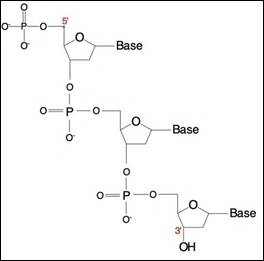
Illustration of how bases are linked in DNA.
167. The two strands of DNA interact with each other via hydrogen bonds between their bases, referred to as Watson-Crick base pairing and twist into a double helix. Adenine generally pairs with thymine (where two hydrogen bonds are formed) and guanine with cytosine (where three hydrogen bonds are formed) as illustrated below:
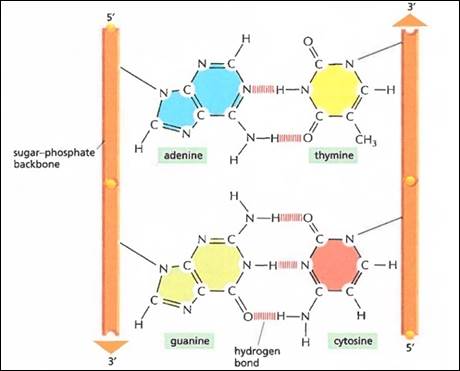
Illustration of adenine pairing with thymine and guanine pairing with cytosine (Alberts et al, Molecular Biology of the Cell (2008) 5th edition ("Alberts"), Fig 4-4).
168. The DNA double helix is further stabilised by interactions between bases known as "base stacking" interactions. These are individually weak interactions, consisting of (i) hydrophobic stacking of the bases on the inside of the helix and (ii) small, short distance van der Waals forces between them. The large number of these low energy interactions in a DNA molecule helps to stabilise the structure of the helix.
169. The coiling of the two strands around each other creates two grooves in the double helix: the wider groove is called the major groove and the smaller groove is called the minor groove.
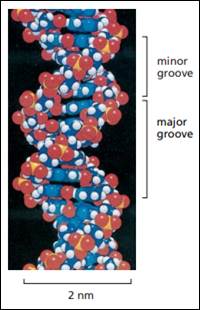
Space filling model of 1.5 turns of the DNA double helix.
170. The outside of the double helix presents DNA sequence information which proteins that regulate the function of DNA can recognise without having to open the double helix. The edges of the base pairs are exposed at the surface of the molecule, presenting a pattern in both the major and minor grooves of hydrogen bond donors and acceptors, and hydrophobic patches for proteins to recognise.
171. Like DNA, RNA (ribonucleic acid) is a polymer comprised of a sugar-phosphate backbone attached to nucleobases. However, RNA differs from DNA in three respects:
172. First, the sugar moiety is hydroxylated at the 2' position in RNA and is therefore ribose instead of deoxyribose, as illustrated below:
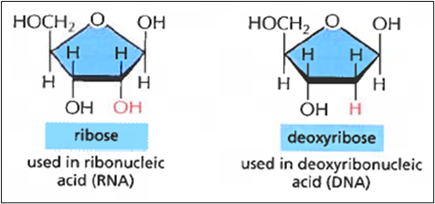
Structures of ribose and deoxyribose (Alberts, Fig 6-4).
173. Second, thymine is replaced by uracil (U) which base pairs with adenine in the same way as thymine does in DNA:
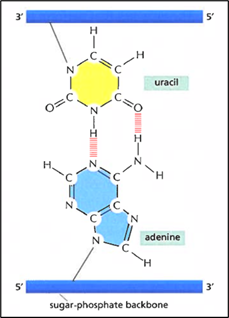
Illustration of uracil base pairing with adenine (Alberts, Fig 6-5).
174. Third, whereas DNA is predominantly double stranded, RNA is generally single-stranded, although it is capable of forming intramolecular double-stranded regions.
175. The capacity of RNA to form complex structures (known as secondary and tertiary structures) means that double-stranded RNA ("dsRNA") can form sections of double helix in the same way as DNA. The final shape of the RNA molecule enables it to recognise other molecules by binding to them selectively.
176. Cells produce several types of RNA, as summarised in the table below.

Types of RNA produced in cells.
177. DNA encodes proteins. Generally speaking, each region of DNA that encodes a particular protein is called a gene. The term "gene expression" (or simply "expression") refers to the process by which the genetic information contained in a gene is represented in the form of its corresponding protein. Some genes are not represented in the form of a protein but are nevertheless "expressed", for example genes encoding tRNA or rRNA which are transcribed but not translated. This process involves two discrete steps: transcription (of DNA to RNA) and translation (of RNA to protein). The type of RNA that participates in both steps is called messenger RNA ("mRNA"). The processes of transcription and translation are discussed further below.
178. The final product of many genes, however, is the RNA itself. These non-coding RNAs serve as enzymatic and structural components for various processes in the cell.
179. The most abundant non-coding RNAs are the ribosomal RNAs ("rRNA"), which coalesce with an array of different proteins to form ribosomes. During protein synthesis the ribosome latches onto the end of an mRNA molecule before moving along the sequence, and aligning it with a second type of RNA, known as transfer RNA ("tRNA") which carries each amino acid into place. In this way the amino acids are brought together in the correct sequence to form a protein (see further below).
180. Further sub-types of RNA include non-coding RNAs such as the microRNAs ("miRNAs"), and small interfering RNAs ("siRNAs").
181. miRNAs are short (21-26 nucleotide) RNAs that bind to specific mRNAs through complementary base-pairing and regulate their stability and translation, thereby either reducing protein output or causing the destruction of the mRNA. They are common endogenous regulators in both animals and plants.
182. siRNAs are considered part of a defence mechanism known as RNA interference, which is triggered by the presence of (usually) exogenous (e.g., viral) dsRNA in the cell. The dsRNA is cleaved by the cellular machinery into short (typically 20-24 nucleotide) siRNA fragments which bind with any complementary RNA molecules leading to their rapid destruction. Such siRNAs can be introduced into cells as dsRNAs, or as single-stranded RNAs that fold in a hairpin structure ("shRNA"), which are then processed to active siRNAs within the cell.
183. Transcription proceeds as follows:
i) a region of the DNA double helix is unwound to expose the bases;
ii) one of the two DNA strands serves as a template; and
iii) a new RNA chain (referred to as a "transcript") is constructed with a nucleotide sequence determined by complementary base-pairing with the DNA template. Synthesis of mRNA is effected by enzymes called RNA polymerases.
184. In eukaryotic cells, transcription takes place within the nucleus.
185. mRNA undergoes three types of processing in the cell: "capping" by the addition of a special nucleotide at its 5' end (the "5' cap"), removal of introns (noncoding sequences that interrupt the coding sequences) from the middle of the mRNA molecule to leave only the coding sequences, or exons (known as "splicing"), and generation of the 3' end of the mRNA by cleavage and polyadenylation. The result is shown below:

The structure of eukaryotic mRNA with a cap at the 5' end and a polyA tail at the 3' end.
186. Mature eukaryotic mRNA i.e. mRNA that has the capacity to survive in the cellular environment and be translated into protein, therefore generally includes the following components:
· 5' cap: in eukaryotic cells, a methylated form of guanosine is added to the 5' end of the mRNA via a 5' to 5' triphosphate linkage. This "cap" stabilises the mRNA and enhances translation. When mRNA is transcribed in vitro (see below), it can be capped post-transcriptionally (i.e. after completion of transcription) or alternatively co-transcriptionally by adding an analog containing a 5'-5' triphosphate linkage to the transcription reaction mixture. One such analog is m7GpppN, "m7G" referring to 7-methylguanosine, "ppp" to three phosphate groups and "N" is any nucleotide. If further methyl groups are present at the 2'-O position on the 1st, 2nd and/or 3rd nucleotides of the RNA proper, these are referred to as "Cap-1", "Cap-2", "Cap-3", respectively;
· 5' UTR: the untranslated region ("UTR") upstream of the coding region that recruits the ribosome and contains signals to initiate translation (see below);
· Coding region/sequence: the region of the mRNA translated into protein, also known as the open reading frame or "ORF";
· 3' UTR: the region located downstream of the coding region that regulates the stability of mRNA as well as translation; and
· PolyA tail: mature eukaryotic mRNA is generally polyadenylated, meaning that a chain of predominantly adenine nucleotides is added at the 3' end of the molecule. The resulting "polyA tail" increases the stability of the mRNA in the cytoplasm and promotes its translation. It is usually denoted by An, where n is the approximate number of adenines in the polyA tail. In eukaryotic cells, polyA tails are typically around 100-200 residues in length. In vitro transcribed RNA can be polyadenylated in two ways: either by including a poly(T) tail in the template or with an enzyme called polyadenylate polymerase. In the case of the latter, the length of the polyA tail is difficult to engineer precisely because it is merely a function of how long the polyadenylation reaction is incubated.
187. In addition to the processing described above, some of the individual nucleotides at specific, conserved locations also undergo post-transcriptional modification as part of the intra-cellular RNA maturation process, catalysed by very specific modifying enzymes.
188. Transcription can also be carried out in vitro (known as in vitro transcription or IVT). IVT products, also known as synthetic RNA, are useful therapeutic and research tools and can be delivered to the cytoplasm of cells to induce the translation of the protein(s) they encode. IVT requires a purified DNA template. The DNA template includes the gene of interest together with an upstream sequence (known as a promoter) which is recognised by the RNA polymerase. The DNA template is incubated with an appropriate RNA polymerase and a mixture of NTPs (nucleoside triphosphates, i.e., ATP, CTP, GTP and UTP representing the four nucleotides found in RNA).
189. The production of mRNA by IVT was routine and was facilitated by the commercial availability of kits.
190. With the simple enzymatic components used in IVT, it is possible to make mRNA transcripts ranging in size from less than 30 nt to about 10,000 nt, enabling the production of mRNA encoding large proteins. In contrast, the alternative approach of chemical synthesis (or "oligosynthesis") is known to have an upper limit of around 200 nt, above which errors tend to accumulate. Oligosynthesis is only really suitable for the production of short polynucleotides.
191. Each of the components of mature mRNA listed above must also be replicated during IVT production of synthetic mRNA, to have a functional mRNA molecule that can induce protein production upon delivery into a cell.
192. Translation is the process by which the sequence of nucleotides in an mRNA molecule is converted into the corresponding chain of amino acids which constitutes the protein in question. For this purpose, the mRNA is "read" in the 5' to 3' direction, three nucleotides at a time. Each group of three nucleotides (referred to as a codon) corresponds to a particular amino acid. For example, AAA represents the amino acid lysine, AAC represents asparagine and so on. Translation is carried out by ribosomes in conjunction with transfer RNAs (tRNAs). Ribosomes contain ribosomal RNA (rRNA) and proteins arranged in the form of two sub-units. tRNAs are cloverleaf-shaped molecules which carry designated amino acids into the active site of the ribosome. One arm of the tRNA molecule includes a group of three nucleotides (referred to as an anticodon) which base-pairs with the codon specifying the amino acid in question.
193. Translation is initiated when the subunits of a ribosome assemble on the mRNA molecule towards its 5' end. The ribosome then "scans" for a start codon (AUG) at which point the corresponding tRNA (with a UAC anticodon) is held in position. Another tRNA molecule then repeats the process for the next codon, and enzymes generate a peptide bond between the two amino acids.
194. This process continues until the ribosome encounters a stop codon, at which point the ribosome detaches from the mRNA and the peptide chain is released.
195. In eukaryotic cells, translation takes place outside the nucleus in the cytoplasm or on ribosomes attached to internal membranes.
196. In IVT mRNA, unmodified canonical nucleotides can be replaced with certain modified, non-canonical nucleotides. Pseudouridine (often referred to as Ψ) was known to reduce the immunogenicity of the mRNA when delivered to cells. Also, capping IVT mRNA with analogues containing m7G were known to result in increased protein production and stability (although m7G would be regarded as an addition rather than a replacement).
197. 
A number of modified nucleotides exist in nature, especially in rRNA and tRNA. For example, pseudouridine and 2'-O-methylated nucleosides (both depicted below) are found in eukaryotic rRNA.
Depiction of pseudouridine (Ψ) and 2'-O-methylated nucleosides (Alberts, Fig 6-43).
198.
Eukaryotic tRNAs are a heavily modified subgroup and contain modified nucleotides including pseudouridine and a number of other modifications, as depicted below:
Depiction of modified tRNA (Stryer, Fig 29,25).
199. The extent of post-transcriptional modification, which results in "non-canonical", or modified, nucleotides, depends on RNA subtype.
200. The Skilled Person would know of databases containing modified nucleotides, such as the RNAMD.
201. It was known that m5C was found in CpG motifs in eukaryotic DNA, 2'-O-methylated nucleotides were often found neighbouring the m7G terminal cap, and m7G was part of the 5'-terminal cap in mammalian mRNA.
202.
Pseudouridine was the first naturally occurring modified nucleoside to be discovered and was subsequently shown to be one of the most abundant modified nucleosides found in nature.
Uridine and pseudouridine (Ψ) nucleoside structures with differences circled in red.
203. In 2010, it was possible to obtain a limited number of modified nucleotides (both naturally occurring and artificial) from commercial sources, for example TriLink in California.
204. It was known from Katalin Karikó and Drew Weissman's 2005 paper (see paragraph 247 below) that certain modified nucleosides could be incorporated into IVT mRNA.
205. Nucleic acids have long been recognised as potential therapeutic agents. Throughout most of the 1990s, the main focus was on DNA as a vehicle for delivering recombinant proteins or antigens into cells, either using plasmids or viral vectors. This work continued in the 2000s (and beyond) although the DNA approach was known to be potentially problematic in three respects.
206. First, in order to be transcribed in eukaryotic cells, exogenous DNA needs to enter the nucleus. However, the nuclear envelope only breaks down during cell division which can lead to a significant delay between exposure (transfection or infection) and expression. Lack of access to the nucleus also made this approach unsuitable for non-dividing cells.
207. Second, there was a perceived risk of insertional mutagenesis arising from integration of exogenous DNA into chromosomal DNA.
208. Third, plasmid DNA is amplified in bacteria, with the consequence that CpG motifs are usually unmethylated (unlike mammalian DNA in which such motifs are usually methylated). Unmethylated CpG motifs are also common in viral DNA. DNA containing unmethylated CpG motifs was known to be highly immunogenic in mammalian cells, acting via a receptor known as TLR 9 (Toll-like receptor 9).
209. siRNA and shRNA were widely employed to study gene function through RNA interference ("RNAi"). Using these small molecules and the cell's own machinery, researchers could selectively silence or knockdown specific genes of interest, hence the name RNAi, allowing them to investigate the consequences of gene loss on cellular processes and phenotype.
210. Similarly, antisense oligonucleotides are short, single stranded RNA (or DNA) molecules designed to bind through base-pairing to a complementary sequence on a specific mRNA molecule. They were known to modulate gene expression by inhibiting translation or promoting degradation of the targeted mRNA and to have potential applications involving the modulation of gene expression.
211. mRNA encoding reporter genes, such as green fluorescent protein ("GFP") or luciferase ("luc"), could be delivered for visualising and quantifying gene expression and for studies of the efficacy of mRNA platforms.
212. mRNA delivery was also employed in cellular reprogramming studies. For example, the introduction of transcription factor mRNAs was able to facilitate the conversion of somatic cells into induced pluripotent stem cells.
213. RNA aptamers (short ssRNA molecules) could be delivered to bind specifically to target proteins.
214. RNA was also known to have a variety of potential therapeutic applications, with a primary focus on mRNA, siRNA, and miRNA. RNA-based therapeutics were gaining prominence due to their ability to modulate gene expression and cellular functions, providing a versatile platform for addressing various diseases.
215. RNA delivery was a key strategy in gene therapy, where the goal was to correct or replace faulty genes. mRNA-based gene therapy was considered particularly promising in monogenetic disorders (disorders affecting only a single gene), such as in cystic fibrosis where the cystic fibrosis transmembrane regulator protein ("CFTR") gene is mutated so the protein is dysfunctional, unstable or not produced.
216. siRNA and miRNA were being investigated to modulate gene expression (siRNA being able to silence specific genes by targeting and degrading their mRNA, and miRNA known to regulate gene expression post-transcriptionally). Antisense RNA ("asRNA") was seen as a promising way of supressing the expression of endogenous or viral genes by hybridising with natural mRNA. It had been investigated for the treatment of genetic disorders, degenerative conditions and certain types of cancer, but had been found to be less efficient than siRNA and impractical due to the difficulty of delivering the large concentrations of asRNA required.
217. RNA-based therapeutics were also being explored for cancer treatment. mRNA vaccines were being used to encode tumour-specific antigens, harnessing the immune system to recognise and target cancer cells. Additionally, siRNA and miRNA were being employed to modulate the expression of genes involved in cancer progression or immune evasion.
218. Whether for research purposes or in the context of therapeutic applications, the Skilled Person would have known that the use of RNA in vivo would require its intact delivery to target cells of interest and its survival long enough to achieve its intended function.
219. Various types of exogenous RNA were known to be labile, prone to degradation by extra- and intracellular nucleases and recognised by the innate immune system.
220. By 2010, it had been known for many years that mRNA had potential as a therapeutic agent. Although IVT mRNA might only deliver transient expression (and therefore require repeated administration), it had certain advantages over DNA:
i) delivery to cytosol – (unlike DNA-based approaches) IVT mRNA only needs to reach the cytosol, rather than the nucleus. Expression can therefore be rapid and can work in both dividing and non-dividing cells;
ii) no insertional mutagenesis – since mRNA cannot integrate into the genome, there is no risk of insertional mutagenesis; and
iii) ease of synthesis and expression – transcribing mRNAs in vitro is relatively straightforward and can be carried out without the need for bacterial amplification.
221. However, IVT mRNA had certain well-known characteristics that were relevant to its use and there remained interest in potential improvements to optimise at least these characteristics:
i) short half-life in the cell cytosol - mRNA is degraded by enzymes called RNases, which are ubiquitous in the environment and in vivo. mRNA delivered in vivo is liable to be degraded by RNases even before it reaches a cell. Various features of mRNA sequence design were well known to impact intra-cellular half-life, including the 5' cap, 3' UTR and polyA tails. One meaning of the word "stability" in the context of mRNA is "biological stability" i.e., length of mRNA survival in vivo, and within the cell - which the Skilled Person would have considered to be closely linked to translational efficiency, with longer survival in the cell providing a correspondingly longer opportunity for translation.
ii) the need for efficient delivery – in order to have its desired pharmacological effect, any nucleic acid needs to cross the cell membrane in order to enter the cell. The chemical structure of mRNA, and its interaction with the structure of a membrane, make this a challenge. Cationic lipid-based delivery vehicles had improved the intracellular delivery of mRNA in vivo and commercially available reagents were available for in vitro transfection; and
iii) its immunogenicity - the work of Karikó and colleagues had shown the potential of certain nucleoside modifications to substantially reduce or eliminate the immunogenicity of mRNA (see further below).
222. mRNA was known to be a fragile molecule and susceptible to degradation and so handling it in the lab requires extensive preparation and cleaning regimes.
223. Work had been undertaken seeking to improve mRNA's prospects, including as a therapeutic agent. In outline, strategies were:
i) improving transfection;
ii) use of improved 5' caps;
iii) polyA tails;
iv) untranslated regions;
v) reducing immunogenicity; and
vi) codon optimisation.
224. To achieve the desired effect, IVT mRNA needs to enter the target cell. However, all mammalian cells are encapsulated in a membrane, which only permits certain molecules to pass through via specific routes.
225. Nucleic acid molecules carry a negative charge about their surface, and that is generally repelled by the electron-rich surface of the membrane.
226. Various reagents and techniques for delivering nucleic acids (including mRNA) had been investigated by 2010.
227. IVT mRNA can be capped with m7GpppG. However, this analog can initiate transcription in either orientation (m7G(5')pppG... or G(5')pppm7G...) but only the former provides a functional cap. Before 2010 so-called "anti-reverse-cap analogs" ("ARCAs"), which can only initiate transcription in the 'correct' orientation, were evaluated to address this.
228. As explained above, there were strategies for adding a polyA tail to IVT mRNA. The tail can be encoded by the template. Alternatively, the transcribed mRNA can be "tailed" using recombinant polyadenylate polymerase. The template approach is more controllable: the number of A residues on the tail is then pre-set, whereas the enzymatic approach yields a population of different tail lengths. For this reason, the one-step approach was generally preferred in 2010.
229. By 2010 it was well known that UTRs can regulate gene expression in important ways.
230. By altering the UTRs that are incorporated in an IVT mRNA, it is possible to improve both its stability and its translational efficiency (and therefore, ultimately, the amount of protein expression).
231. In 2010 the Skilled Person would have known that Karikó / Weissman's group had demonstrated that replacing uridine with pseudouridine reduced the activation of TLR3, TLR7 and TLR8 receptors in cultured cells (see further below concerning TLRs).
232. By 2010, it was known that the efficiency of translation could be improved by choosing carefully between the alternative codons for a given amino acid.
233. As the first line of defence against invading pathogens, components of innate immunity are mostly present before the onset of infection. They are not specific to particular pathogens and rely on the recognition of classes of molecules frequently encountered in pathogens, such as nucleic acids, or other non-specific mechanisms (like mucosal barriers).
234. Part of the innate immune system relies on pattern recognition molecules that can recognise a given class of molecules. This renders the system capable of immediately recognising and initiating immune responses against invaders displaying molecular markers characteristic of microbes.
235. Although innate immune cells, including macrophages and dendritic cells, play important roles, other 'non-professional' cells also contribute to innate immunity.
236. Pattern recognition receptors ("PRRs") are responsible for sensing the presence of microorganisms. They do this by recognising structures conserved among microbial species, including structures on nucleic acids, which are called pathogen-associated molecular patterns ("PAMPs").
237. As at the EP 949 Priority Date, several different classes of PRR families had been identified and studied. These included the transmembrane proteins known as Toll-like receptors ("TLRs"). These PRRs are expressed not only in macrophages and dendritic cells but also in the various other non-professional immune cells mentioned above, although the pattern of expression may vary.
238. The sensing of PAMPs by PRRs upregulates the transcription of genes involved in inflammatory responses. These genes encode various proteins involved in the modulation of PRR signalling including inflammatory cytokines such as tumour necrosis factor ("TNF"), interleukin ("IL"), and type I interferons ("IFNs").
239. The cytokines are proteins that regulate protein production and cell death in inflammatory tissues.
240. DNA and RNA have long been known to stimulate the mammalian innate immune system by triggering certain receptors, including several TLRs. "Foreign" nucleic acids such as dsRNA originating from viruses and bacteria are potent activators of this system (see above).
241. The innate immune system represents a challenge for the use of exogenous RNA in cell lines or in vivo, for example siRNA for RNAi and mRNA for the induction of protein production. Synthetic RNA can activate the innate immune system in similar ways as foreign nucleic acids originating from pathogens, triggering an inflammatory response and a signalling cascade that can reduce protein expression or even destroy transfected cells entirely.
242. In particular, TLR3 was known to recognise dsRNA, which is produced during the replication of many viruses as either an intermediate in RNA replication or the double-stranded RNA genome. Similarly, TLR7 and TLR8 were known to be activated by viral and synthetic single-stranded RNA.
243. TLR9 was known to recognise DNA, particularly its CpG motif, consisting of the unmethylated dinucleotide CpG flanked by two 5' purine residues and two 3' pyrimidines. When the CpG motif does occur it is mostly methylated.
244. Methyl groups are found at the 2' position of the ribose in 2'-O-methylnucleotides and were seen in nature in eukaryotic RNAs including tRNA and rRNA.
245. Katalin Karikó and Drew Weissman had published papers in the mid-2000s on the immunogenicity of unmodified and nucleoside modified IVT mRNA. The finding that unmodified IVT mRNA stimulated the innate immune system by activating TLR3, TLR7 and TLR8, was well known and generally accepted by 2010.
246. The Weissman/Karikó group established that certain naturally occurring nucleotide modifications played a role in the recognition of mammalian mRNA by the innate immune system and that by incorporating certain modified nucleosides in IVT mRNA it was possible to overcome its immunogenicity.
247. "Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA" ("Karikó 2005") is a paper from the Weissman/Karikó group describing a series of experiments that examined the effect of nucleotide modification on the immunogenicity of IVT mRNA. It was published in the journal Immunity and was an important and widely read publication. Its full reference is Immunity 2005; 23: 165-175.
248. The group showed that pseudouridine could reduce the activation of TLR3, TLR7 and TLR8 in cultured cells thereby diminishing the immunogenicity of IVT mRNA.
249. The parties identified four disputed areas which are set out below. Pfizer/BioNTech contended that the only dispute of relevance was that relating to the RNAMD (see iii below).
i) The extent of the Skilled Person's knowledge of specific naturally occurring modified nucleosides/nucleotides.
ii) The effect of such nucleosides on mRNA stability, protein production and immunogenicity.
iii) The familiarity of the Skilled Person with the RNAMD.
iv) The extent to which the experimental work and the specific modifications tested in Karikó 2005 were CGK.
250. Moderna conceded point (iii) following the oral evidence and in my view it was right to do so. This means that the skilled person would know that the RNAMD existed, its purpose, the nature of its contents, and that it was searchable. They would not know its contents by heart but would know how to access it when they needed to.
251. On points (i), (ii) and (iv) Moderna agreed with the following (with a minor deletion) as CGK as stated in Pfizer/BioNTech's closing skeleton:
i) The skilled person would take away from Karikó 2005, and it would be CGK, that certain naturally occurring modified nucleotides suppress the capacity of RNA to activate the innate immune response, and suppression is proportional to the number of modifications.
ii) It was known that some nucleotides which were methylated had reduced immunogenicity, like m7G of the 5' cap and m5C. This knowledge came from Karikó 2005 and also from earlier knowledge about m7G.
iv) The skilled person would recall that it was not just Ψ that was tested in Karikó 2005, other nucleotides were also tested and it would be very easy to go back to the paper and look up which nucleotides had been tested.
252. Kormann et al, "Expression of therapeutic proteins after delivery of chemically modified mRNA in mice", Nature Biotechnology; received 5 October 2010 and published online 9 January 2011 ("Kormann 2011") was co-authored by Prof Rosenecker. It is post-priority so not part of the CGK but it was relied on by the parties on a number of topics so I summarise it here for convenience.
253. The abstract provides a useful summary (footnotes removed):
Current viral vectors for gene therapy are associated with serious safety concerns, including leukemogenesis, and nonviral vectors are limited by low gene transfer efficiency. Here we investigate the therapeutic utility of chemically modified mRNA as an alternative to DNA-based gene therapy. A combination of nucleotide modifications abrogates mRNA interaction with Toll-like receptor (TLR)3, TLR7, TLR8 and retinoid-inducible gene I (RIG-I), resulting in low immunogenicity and higher stability in mice. A single intramuscular injection of modified murine erythropoietin mRNA raises the average hematocrit in mice from 51.5% to 64.2% after 28 days. In a mouse model of a lethal congenital lung disease caused by a lack of surfactant protein B (SP-B), twice weekly local application of an aerosol of modified SPB mRNA to the lung restored 71% of the wild-type SP-B expression, and treated mice survived until the predetermined end of the study after 28 days.
254. The modifications tested were m6A, ΨU, s2U and m5C (along with unmodified mRNA and an s2U and m5C combination). 25% and 50% modifications were tested. The authors reported that they found "replacement of only 25% of uridine and cytidine with 2-thiouridine and 5-methyl-cytidine synergistically decreased mRNA binding to pattern recognition receptors, such as TLR3, TLR7, TLR8 and RIG-I, in human peripheral blood mononuclear cells (PBMCs)."
255. Pfizer/BioNTech sought to use Kormann 2011 to attack Professor Rosenecker's suitability as the RNA Biologist expert witness in this case. Professor Rosenecker said that the decision made by the Kormann 2011 team in relation to the modified nucleosides to test was suggested by Michael Kormann and Carsten Rudolph, and that he "just accepted it". I address this criticism in paragraph 43 above. It was also relevant to the secondary evidence case put forward by Moderna and I return to it there.
256. The parties' rival positions were:
i) Moderna and Prof Rosenecker said that EP949 is directed to a scientist with a good understanding of the biology of RNA who is working on, or has an interest in, developing mRNA for the purposes of transcript therapy;
ii) Pfizer/BioNTech and Dr Enright said that EP949 is directed to an RNA biologist who is interested in using RNA for research, whether fundamental or applied to therapeutic purposes.
257. I will deal with this by applying the approach indicated in the authorities referred to above in the section beginning at paragraph 103, which begins with identifying the problem to be solved. At one level the parties agreed on this. Moderna's closing skeleton stated:
The starting point is the problem sought to be solved by the invention. As to that, there was agreement; at T1/p115/21-24 Dr Enright agreed with Professor Rosenecker's formulation of the problem sought to be solved by the invention of EP949, given in his first report (D.1/1) at §314:
Q. The problem which the invention aims to solve is how to
increase the translation of mRNA and also reduce the
immunogenicity?
A. Yes, I would guess that that is true.
While Pfizer/BioNTech's closing skeleton stated:
It is common ground that the problem that the Patent aims to solve is the problem of increasing the intracellular translation and reducing the immunogenicity of exogenous RNA (Rosenecker 1 §314; Enright XX T1/115/11-24).
258. These formulations amount to the same thing (leaving aside a nuanced difference between mRNA and exogenous RNA which is not relevant to this discussion). Counsel for Pfizer/BioNTech said that there was a qualification because immunogenicity was already addressed in Karikó 2005. But the problem was to achieve both increased translation and reduced immunogenicity at once, so I reject this, although it is not important.
259. This agreement identifies the problem at a scientific and somewhat conceptual level but leaves open the question of why it mattered, and to what practical end a solution to it could be put. So the issue still remains of whether the skilled person would be someone working on using mRNA for transcript therapy (Moderna) or someone who also would have an interest in fundamental research (Pfizer/BioNTech). I am not bound to choose between the parties' two formulations and indeed I go on below to reject both, although the upshot is more in favour of Moderna.
260. As I identify in addressing the specification of EP949, the practical application of what it teaches is not limited to transcript therapy. It also covers immunotherapy and direct vaccination, as well as non-therapeutic uses. I need not go into the details, but Counsel for Pfizer/BioNTech established in cross-examination that there were real teams in the following relevant areas where a solution to the problem could be useful (but it was not suggested and would be unreal for any of those teams to be working in all of them, and indeed I think most if not all were only active in one):
i) Cellular reprogramming studies;
ii) Immunotherapy;
iii) Direct vaccination;
iv) Studying gene expression and the efficacy of RNA platforms;
v) Studying mechanisms of translation and immune response;
vi) Studies on zinc finger nuclease technology;
vii) Neuroscience research;
viii) Developmental research; and
ix) Gene (or protein) replacement therapy.
261. So Moderna is wrong in seeking to define the skilled person as being someone working on, specifically, transcript therapy. They could be working in any of those fields. Pfizer/BioNTech described some of the above fields as "study" or "research". That tended to favour its argument that Dr Enright was close(r) to those teams, but I think it is not a fair way of looking at things. The above teams were looking for practical results and I do not think an overview of them supports the position that the correct field was one of pure research, whatever its scope.
262. A further reason to reject Moderna's narrow definition of the skilled person, as Pfizer/BioNTech submitted and as I accept, is that the claims of EP949 are not limited to therapy.
263. I therefore identify the skilled person as being someone with a knowledge of RNA biology, with a practical interest in improving the use of mRNA in relation to translation and immunogenicity in any of the fields above.
264. That means that there was not any real team corresponding to the notional skilled person in the breadth of their interest, in the sense that no team covered so much ground. It also means that it would have been impossible for Moderna, or indeed either side, to call a single witness who in fact matched that breadth of interest. Pfizer/BioNTech criticised Prof Rosenecker for identifying the skilled person as, and being, someone interested only in transcript therapy when EP949 goes broader, but if Moderna had called someone whose interest was in mRNA for vaccines Pfizer/BioNTech would have made the same point.
265. I think it is unusual but not unprincipled to identify the skilled person as being someone with a practical interest in the use of mRNA where translation and immunogenicity were relevant, even though in the real world the work of any given individual would inevitably be on only a subset of that broader field. This means that it would be open to anyone challenging the validity of EP949 to show that it was obvious to a skilled person working on mRNA vaccines, or on mRNA immunotherapy, or on mRNA for stem cell development. Had Pfizer/BioNTech called an expert from any of these subfields, had the difference between the subfields mattered, and had Prof Rosenecker not been able to put himself in the position of someone in such other subfield, then Moderna's position might have been quite difficult. But that is not what has happened: Pfizer/BioNTech have called an expert who is not from any of the subfields but rather a pure, basic scientist.
266. I also think that in the present case there is an important interface between the argument over the skilled addressee of EP949 and the argument over the right approach to obviousness. Pfizer/BioNTech's argument is that the skilled addressee is a basic scientist interested in fundamental research. The downstream effect of the contention is problematic: it founds Pfizer/BioNTech's submission that it would be obvious to try a variety of Ψ modifications, including m1Ψ, without any particular practical goal in mind, without any concrete expectation of success (indeed with a willingness to accept null results as a success in the sense of providing information), and without understanding why Ψ itself achieved what it did in the prior art. This is not a proper approach to obviousness. I also think it is not the proper approach to the skilled person. The skilled person has a practical interest in the application of an invention, and even if that is not an absolute rule, it is a conclusion that is justified in the present case where EP949 identifies a range of practical applications. The fact that they might be deployed in research in a practical way does not detract from this. I bear in mind that the relevant field may be a research field or a field of manufacture (Illumina at [66]) but patents are nonetheless addressed to readers with a practical interest (e.g. MedImmune at [77]).
267. EP949 is entitled "Ribonucleic acids containing N1-methyl-pseudouracils and uses thereof". For the most part, the parties agreed on the disclosure of EP949, with some minor exceptions which I identify below.
268. In the "Background" section, [0002] states:
[0002] Naturally occurring RNAs are synthesized from four basic ribonucleotides: ATP, CTP, UTP and GTP, but may contain post-transcriptionally modified nucleotides. Further, approximately one hundred different nucleoside modifications have been identified in RNA (Rozenski, J, Crain, P, and McCloskey, J. (1999). The RNA Modification Database: 1999 update. Nucl Acids Res 27: 196-197). The role of nucleoside modifications on the immuno-stimulatory potential, stability, and on the translation efficiency of RNA, and the consequent benefits to this for enhancing protein expression and producing therapeutics however, is unclear.
269. Dr Enright and Professor Rosenecker agreed with this, subject to the CGK of the work described in Karikó 2005.
270. The "Background" section continues in [0003] and states that there are "multiple problems with prior methodologies of effecting protein expression", specifically the use of DNA. The problems include alterations and/or damage to the host cell genomic DNA and the need for multiple processing steps which creates a time lag before the protein of interest can be generated. At [0004] it is stated that "There is a need in the art for biological modalities to address the modulation of intracellular translation of nucleic acids." [0005] cites UPenn.
271. In the "Summary" section, [0008] and [0009] state:
[0008] The present disclosure relates, inter alia, to modified nucleosides, modified nucleotides, and modified nucleic acids which can exhibit a reduced innate immune response when introduced into a population of cells, both in vivo and ex vivo. Further, these modified nucleosides, modified nucleotides, and modified nucleic acids described herein can disrupt binding of a major groove interacting partner with the nucleic acid. Because of the reduced immunogenicity and the decrease in major groove interactions, these modified nucleosides, modified nucleotides, and modified nucleic acids can be more efficient during protein production than, e.g., unmodified nucleic acids.
[0009] Thus, the present disclosure relates to compounds comprising nucleotides that can disrupt binding of a major groove binding partner with a nucleic acid, wherein the nucleotide has decreased binding affinity to the major groove binding partner.
272. EP949 defines "major groove interacting partner" at [0128] by reference to ligands which interact with the major groove face of a nucleotide or nucleic acid. It teaches that RNA ligands comprising modified nucleotides or nucleic acids which decrease interactions with major groove binding partners "therefore decrease an innate immune response, or expression and secretion of pro-inflammatory cytokines, or both."
273. The "Detailed Description" section starts at [0020]:
[0020] The present disclosure relates, inter alia, to modified nucleosides, modified nucleotides, and modified nucleic acids that exhibit a reduced innate immune response when introduced into a population of cells. The modified nucleosides, modified nucleotides, and modified nucleic acids can be chemically modified on the major groove face, thereby disrupting major groove binding partner interactions, which cause innate immune responses.
[0021] In general, exogenous unmodified nucleic acids, particularly viral nucleic acids, introduced into cells induce an innate immune response, resulting in cytokine and interferon (IFN) production and cell death. However, it is of great interest for therapeutics, diagnostics, reagents and for biological assays to deliver a nucleic acid, e.g., a ribonucleic acid (RNA) inside a cell, either in vivo or ex vivo, such as to cause intracellular translation of the nucleic acid and production of the encoded protein. Of particular importance is the delivery and function of a non-integrative nucleic acid, as nucleic acids characterized by integration into a target cell are generally imprecise in their expression levels, deleteriously transferable to progeny and neighbor cells, and suffer from the substantial risk of causing mutation. Described herein in part are nucleic acids encoding useful polypeptides capable of modulating a cell's function and/or activity, and methods of making and using these nucleic acids and polypeptides. As described herein, these nucleic acids are capable of reducing the innate immune activity of a population of cells into which they are introduced, thus increasing the efficiency of protein production in that cell population. Further, one or more additional advantageous activities and/or properties of the nucleic acids and proteins of the present disclosure are described.
[0022] Further, the modified nucleosides, modified nucleotides, and modified nucleic acids described herein can be modified on the major groove face. These major groove modifications can allow for alterations, e.g. a decrease, in the interaction of the modified nucleosides, modified nucleotides, and modified nucleic acids with a binding groove partner.
[0023] Accordingly, in a first aspect, the present disclosure describes compounds comprising a nucleotide that can disrupts binding of a major groove interacting, e.g. binding, partner with a nucleic acid, wherein the nucleotide has decreased binding affinity to major groove interacting, e.g. binding, partners.
274. [0021] in particular makes clear that EP949 is not concerned only with therapy and is one thing that supports my view on the skilled person.
275. A key section of the description is the section entitled "Uses of Modified Nucleic Acids" at [0161] to [0179]. This section starts with the sub-heading "Therapeutic Agents". Paragraphs [0161] and [0162] state:
[0161] The modified mRNA of the invention and the proteins translated therefrom can be used as therapeutic agents. For example, a modified mRNA of the invention can be administered to a subject, wherein the modified nucleic acid is translated in vivo to produce a therapeutic peptide in the subject. Accordingly, the mRNA can be used in compositions, methods, kits, and reagents for treatment or prevention of disease or conditions in humans and other mammals. The active therapeutic agents of the present disclosure include modified mRNA, cells containing modified mRNA or polypeptides translated from the modified mRNA, polypeptides translated from modified mRNA, and cells contacted with cells containing modified mRNA or polypeptides translated from the modified mRNA.
[0162] Described are combination therapeutics containing one or more modified m RNA containing translatable regions that encode for a protein or proteins that boost a mammalian subject's immunity along with a protein that induces antibody-dependent cellular toxicity. For example, therapeutics containing one or more mRNA that encode trastuzumab and granulocyte-colony stimulating factor (G-CSF). In particular, such combination therapeutics are useful in Her2+ breast cancer patients who develop induced resistance to trastuzumab. (See, e.g., Albrecht, Immunotherapy. 2(6):795-8 (2010)). The mRNA of the invention is useful in methods of inducing translation of a recombinant polypeptide in a cell population. Such translation can be in vivo, ex vivo, in culture, or in vitro. The cell population is contacted with an effective amount of a composition containing the mRNA encoding the recombinant polypeptide. The population is contacted under conditions such that the mRNA is localized into one or more cells of the cell population and the recombinant polypeptide is translated in the cell from the nucleic acid.
276. Under the next sub-heading, "Therapeutics for diseases and conditions", [0171] states:
[0171] The mRNA of the invention as defined in the claims can be used in methods for treating or preventing a symptom of diseases characterized by missing or aberrant protein activity, by replacing the missing protein activity or overcoming the aberrant protein activity. Because of the rapid initiation of protein production following introduction of modified mRNAs, as compared to viral DNA vectors, the compounds of the present disclosure are particularly advantageous in treating acute diseases such as sepsis, stroke, and myocardial infarction. Moreover, the lack of transcriptional regulation of the modified mRNAs of the present disclosure is advantageous in that accurate titration of protein production is achievable.
277. In the next paragraph, [0172], it suggests different conditions for which the mRNA of the invention could be used in a method of treatment, including cystic fibrosis.
278. There are further sub-sections on "Methods of cellular nucleic acid delivery", "Targeting Moieties" and "Permanent Gene Expression Silencing" and sections on "Pharmaceutical Compositions" and "Kits" before the examples.
279. EP949 has seven examples and provides data for Examples 1 to 4 only.
280. Example 1 describes the synthesis of modified mRNAs using standard laboratory methods and materials for in vitro transcription, where the nucleotide mix contained modified nucleotides. The synthesised mRNAs are shown in Table 3. Table 3 shows 31 different modifications. The majority are synthesised with 100% replacement of the canonical nucleotide(s) with their modified counterpart(s), but Chem 51 replaces only 25% of the cytidines and Chem 52 replaces only 25% of the uridines.
281. The success of the transcription reaction was analysed by agarose gel electrophoresis (Figs. 1A and 1B). Results for UV absorbance by NanoDrop testing are provided for a subset of the samples (Figs. 6A to 6L).
282. Example 2 transfects keratinocytes with IVT mRNA encoding human granulocyte-colony stimulating factor (hu-G-CSF). The quantity of G-CSF for each of the modified mRNAs was measured using enzyme-linked immunosorbent assays (ELISA) after 18 hours and is shown in Figs. 2A and 2B. Fig. 2A shows the results of reverse transfection and Fig. 2B shows results of forward transfection.
283. Example 3 provides results from ELISA assays for human TNFα, human IFN-β and human G-CSF secreted from IVT human keratinocyte cells. It was common ground that the absence of statistical analysis makes it difficult to draw quantitative conclusions.
284. Example 4 uses KG-1 and Kasumi-1 (two human myeloblast cell lines that express G-CSF receptors) co-cultivated with reverse-transfected keratinocytes acting as "feeder cells". The keratinocytes were transfected with IVT mRNA encoding hu-G-CSF. Proliferation of the myeloblast cells acts as a marker for biologically active hu-G-CSF secretion.
285. Examples 5 and 6 relate to investigating the effect of modified mRNAs on cellular viability, cytotoxicity and apoptosis. No data are provided and the experts did not place any significance on them.
286. I have been relatively brief about the Examples, and much briefer than the parties were in their written submissions. The reason is that there was a possible dispute about whether EP949 shows that m1Ψ is superior to Ψ, or only superior to the unmodified nucleotide. That is because Pfizer/BioNTech wanted to ensure that Moderna did not seek to argue obviousness on the basis that the former was part of the technical contribution; it said that the contribution was no more than providing an alternative to Ψ, not an improvement. So, it said, a motivation to find an alternative would be good enough.
287. In the event, Moderna did not argue that m1Ψ was shown to be superior to Ψ. I have borne this in mind and address it further below, concluding that given the way the obviousness case was developed it does not make a material difference. I note, however, that Pfizer/BioNTech did not argue that m1Ψ is not a useful improvement over the unmodified nucleotide; it is not an arbitrary choice in the AgrEvo sense.
288. The claims in issue are claims 3 and 5; both of which are said by Moderna to be independently valid and infringed.
289. Claim 3 of EP949 is:
An mRNA wherein 100% of nucleotides comprising uracil in the mRNA are replaced with nucleotides comprising N1-methyl-pseudouridine.
290. Claim 5 of EP949 is:
An mRNA according to any of claims 3 or 4 comprising a polyA tail.
291. I will deal first with UPenn and then with Karikó 2008. This is not chronological order, and the citations have to be considered separately, but UPenn is Pfizer/BioNTech's stronger case. Most of the themes and arguments, and all the key ones, are exactly the same; a lot of the oral evidence was in the context of Karikó 2008 but applies identically to UPenn.
292. It was clear on the evidence that both citations would be recognised as very high quality work and the skilled person would recognise that the authors were scientists of great repute in the field. The fact that UPenn is a patent filing and contains a fair bit of patent language and boilerplate (whereas Karikó 2008 is a peer reviewed scientific publication) would not undermine this assessment of the technical contents. So the skilled person would take both documents' teachings seriously.
293. Another reason for dealing with UPenn first is that Pfizer/BioNTech relies on it as an anticipation. I therefore set out the key passages relied on for that attack relatively fully; I summarise other findings in shorter form. The fact that UPenn is relied on for anticipation makes it convenient to take this approach in this judgment, but it has no relevance to the assessment of inventive step over Karikó 2008.
294. UPenn is an international patent application with a publication date of 1 March 2007. The inventors are Katalin Karikó and Drew Weissman.
295. The title is "RNA Containing Modified Nucleosides and Methods of Use Thereof".
296. Paragraph [001], which relates to the Field of Invention, states:
[001] This invention provides RNA, oligoribonucleotide, and polyribonucleotide molecules comprising pseudouridine or a modified nucleoside, gene therapy vectors comprising same, methods of synthesizing same, and methods for gene replacement, gene therapy, gene transcription silencing, and the delivery of therapeutic proteins to tissue in vivo, comprising the molecules. The present invention also provides methods of reducing the immunogenicity of RNA, oligoribonucleotide, and polyribonucleotide molecules.
297. Paragraph [003] under the heading "Summary of the Invention" is as follows:
[003] This invention provides RNA, oligoribonucleotide, and polyribonucleotide molecules comprising pseudouridine or a modified nucleoside, gene therapy vectors comprising same, gene therapy methods and gene transcription silencing methods comprising same, methods of reducing an immunogenicity of same, and methods of synthesizing same.
298. The main relevant paragraphs from UPenn thereafter are as follows (I have not quoted everything to which reference was made and I have not attempted to narrate the details of the experimental work other than those particularly relied on for anticipation - I give a high level overview at the end of this section).
[004] In one embodiment, the present invention provides a messenger RNA comprising a pseudouridine residue.
[006] In another embodiment, the present invention provides an in vitro-transcribed RNA molecule, comprising a pseudouridine or a modified nucleoside.
[0051] In another embodiment, the RNA, oligoribonucleotide, or polyribonucleotide molecule further comprises a poly-A tail. In another embodiment, the RNA, oligoribonucleotide, or polyribonucleotide molecule does not comprise a poly-A tail. Each possibility represents a separate embodiment of the present invention.
[0056] "Pseudouridine" refers, in another embodiment, to m1acp3Ψ (1-methyl-3-(3-amino-3-carboxypropyl) pseudouridine. In another embodiment, the term refers to m1Ψ (1-methylpseudouridine). In another embodiment, the term refers to Ψm (2'-O-methylpseudouridine. In another embodiment, the term refers to m5D (5-methyldihydrouridine ). In another embodiment, the term refers to m3Ψ (3-methylpseudouridine). In another embodiment, the term refers to a pseudouridine moiety that is not further modified. In another embodiment, the term refers to a monophosphate, diphosphate, or triphosphate of any of the above pseudouridines. In another embodiment, the term refers to any other pseudouridine known in the art. Each possibility represents a separate embodiment of the present invention.
[0069] In another embodiment, the modified nucleoside of methods and compositions of the present invention is m5C (5-methylcytidine). In another embodiment, the modified nucleoside is m5U (5- methyluridine). In another embodiment, the modified nucleoside is m6A (N6-methyladenosine). In another embodiment, the modified nucleoside is s2U (2-thiouridine). In another embodiment, the modified nucleoside is Ψ (pseudouridine). In another embodiment, the modified nucleoside is Um (2'-O-methyluridine).
[0072] In another embodiment, between 0.1 % and 100% of the residues in the RNA, oligoribonucleotide, or polyribonucleotide molecule of methods and compositions of the present invention are modified (e.g. either by the presence of pseudouridine or a modified nucleoside base). In another embodiment, 0.1% of the residues are modified. In another embodiment, 0.2%. In another embodiment, the fraction is 0.3%. In another embodiment, the fraction is 0.4%. In another embodiment, the fraction is 0.5%. In another embodiment, the fraction is 0.6%. In another embodiment, the fraction is 0.8%. In another embodiment, the fraction is 1 %. In another embodiment, the fraction is 1.5%. In another embodiment, the fraction is 2%. In another embodiment, the fraction is 2.5%. In another embodiment, the fraction is 3%. In another embodiment, the fraction is 4%. In another embodiment, the fraction is 5%. In another embodiment, the fraction is 6%. In another embodiment, the fraction is 8%. In another embodiment, the fraction is 10%. In another embodiment, the fraction is 12%. In another embodiment, the fraction is 14%. In another embodiment, the fraction is 16%. In another embodiment, the fraction is 18%. In another embodiment, the fraction is 20%. In another embodiment, the fraction is 25%. In another embodiment, the fraction is 30%. In another embodiment, the fraction is 35%. In another embodiment, the fraction is 40%. In another embodiment, the fraction is 45%. In another embodiment, the fraction is 50%. In another embodiment, the fraction is 60%. In another embodiment, the fraction is 70%. In another embodiment, the fraction is 80%. In another embodiment, the fraction is 90%. In another embodiment, the fraction is 100%.
EXAMPLE 2: IN VITRO SYNTHESIS OF RNA MOLECULES WITH MODIFIED NUCLEOSIDES
MATERIALS AND EXPERIMENTAL METHODS
In vitro-transcribed RNA
[00187] Using in vitro transcription assays (MessageMachine and MegaScript kits; Ambion,) the following long RNAs were generated by T7 RNA polymerase (RNAP) as described (Kariko et al, 1998, Phosphate-enhanced transfection of cationic lipid-complexed mRNA and plasmid DNA. Biochim Biophys Acta 1369, 3 20-334) (Note: the names of templates are indicated in parenthesis; the number in the name of the RNA specifies the length): RNA-1866 (Nde I-linearized pTEVluc) encodes firefly luciferase and a50 nt-long poly A-tail. RNA-1571 (Ssp I-linearized pSVren) encodes Renilla luciferase. RNA-730 (Hind III-linearized pT7T3D-MART-l) encodes the human melanoma antigen MART-1. RNA-713 (EcoR I-linearized pT7T3D-MART-1) corresponds to antisense sequence of MART-1, RNA-497 (Bgl II-linearized pCMV-hTLR3) encodes a partial 5' fragment of hTLR3. Sequences of the RNA molecules are as follows: ...
299. The sequences listed in paragraphs [00188] to [00192] are RNA-1866 (which has a poly A-tail, discussed further below), RNA-1571, RNA-730, RNA-713 (an anti-sense strand i.e. not mRNA) and RNA-497. The Example continues:
[00193] To obtain modified RNA, the transcription reaction was assembled with the replacement of one (or two) of the basic NTPs with the corresponding triphosphate-derivative(s) of the modified nucleotide 5-methylcytidine, 5-methyluridine, 2-thiouridine, N6-methyladenosine or pseudouridine (TriLink, San Diego, CA). In each transcription reaction, all 4 nucleotides or their derivatives were present at 7.5 millimolar (mM) concentration. In selected experiments, as indicated, 6 mM m7GpppG cap analog (New England BioLabs, Beverly, MA) was also included to obtain capped RNA. ORN5 and ORN6 were generated using DNA oligodeoxynucleotide templates and T7 RNAP (Silencer® siRNA construction kit, Ambion).
300. Paragraph [00193] above confirms that the method involves replacement of 100% of the chosen nucleoside, that is where every instance of a given nucleoside (adenosine, guanosine, cytosine or uridine) is replaced with a modified version. This is further confirmed in Example 7 below. The results of Example 2 are provided:
RESULTS
[00194] To further test the effect of nucleoside modifications on immunogenicity, an in vitro system was developed for producing RNA molecules with pseudouridine or modified nucleosides. In vitro transcription reactions were performed in which 1 or 2 of the 4 nucleotide triphosphates (NTP) were substituted with a corresponding nucleoside-modified NTP. Several sets of RNA with different primary sequences ranging in length between 0.7-1.9 kb, and containing either none, 1 or 2 types of modified nucleosides were transcribed. Modified RNAs were indistinguishable from their non-modified counterparts in their mobility in denaturing gel electrophoresis, showing that they were intact and otherwise unmodified (Figure 2A). This procedure worked efficiently with any of T7, SP6, and T3 phage polymerases, and therefore is generalizable to a wide variety of RNA polymerases.
[00195] These findings provide a novel in vitro system for production of RNA molecules with modified nucleosides.
...
EXAMPLE 7: SUPPRESSION OF RNA-MEDIATED IMMUNE STIMULATION IS PROPORTIONAL TO THE NUMBER OF MODIFIED NUCLEOSIDES PRESENT IN RNA
...
[00212] Most of the nucleoside-modified RNA utilized thus far contained one type of modification occurring in approximately 25% of the total nucleotides in the RNA (e.g. all the uridine bases). To define the minimal frequency of particular modified nucleosides that is sufficient to reduce immunogenicity under the conditions utilized herein, RNA molecules with limited numbers of modified nucleosides were generated. In the first set of experiments, RNA was transcribed in vitro in the presence of varying ratios of m6A, Ψ or m5C to their corresponding unmodified NTPs. The amount of incorporation of modified nucleoside phosphates into RNA was expected to be proportional to the ratio contained in the transcription reaction, since RNA yields obtained with T7 RNAP showed the enzyme utilizes NTPs of m6A, Ψ or m5C almost as efficiently as the basic NTPs. To confirm this expectation, RNA transcribed in the presence of UTP:Ψ in a 50:50 ratio was digested and found to contain UMP and Ψ in a nearly 50:50 ratio (Figure 5A).
301. The first sentence of paragraph [00212] above supports the understanding that Example 2 teaches 100% replacement of the chosen nucleoside.
302. Example 31 was key to the dispute between the parties:
EXAMPLE 31: TESTING THE EFFECT OF ADDITIONAL NUCLEOSIDE MODIFICATIONS ON RNA IMMUNOGENICITY AND EFFICIENCY OF TRANSLATION
[00290] Additional nucleoside modifications are introduced into in vitro-transcribed RNA, using the methods described above in Examples 2 and 7, and their effects on immunogenicity translation efficiency are tested as described in Examples 1-8 and 9-15, respectively. Certain additional modifications are found to decrease immunogenicity and enhance translation. These modifications are additional embodiments of methods and compositions of the present invention.
[00291] Modifications tested include, e.g.: m1A; m2A; Am; ms2m6A; i6A; ms2i6A; io6A; ms2io6A; g6A; t6A; ms2t6A; m6t6A; hn6A; ms2hn6A; Ar(p); I; m1I; m1Im; m3C; Cm; s2C; ac4C; f5C; m5Cm; ac4Cm; k2C; m1G; m2G; m7G; Gm; m22G; m2Gm; m22Gm; Gr(p); yW; o2yW; OHyW; OHyW*; imG; mimG; Q; oQ; galQ; manQ; preQ0; preQ1; G+; D; m5Um; m1Ψ; Ψ m; s4U; m5s2U; s2Um; acp3U; ho5U; mo5U; cmo5U; mcmo5U; chm5U; mchm5U; mcm5U; mcm5Um; mcm5s2U; nm5s2U; mnm5U; mnm5s2U; mnm5se2U; ncm5U; ncm5Um; cmnm5U; cmnm5Um; cmnm5s2U; m62A; Im; m4C; m4Cm; hm5C; m3U; m1acp3' Ψ; cm5U; m6Am; m62Am; m2,7G; m2,2,7G; m3Um; m5D; m3 Ψ; f5Cm; m1Gm; m1Am;πm5U; πm5s2U; imG-14; imG2; and ac6A.
303. Returning to the experimental work, there are six modified nucleosides for which experimental data are given, listed in [0069]: m5C (5-methylcytidine), m5U (5-methyluridine), m6A (N6-methyladenosine), s2U (2-thiouridine), Ψ (pseudouridine), and Um (2'-O-methyluridine). They vary in structure quite widely. There are in vitro experiments, experiments in cultured cells, and in vivo experiments. The only modification taken into the in vivo experiments is Ψ.
304. The key message of UPenn about the experiments is contained in paragraphs [00241] and [00244]-[00246], in the context of Examples 13-14:
[00241] Thus, pseudouridine modification increases RNA translation efficiency in vitro, in cultured cells, and in vivo- in multiple animal models and by multiple routes of administration, showing its widespread application as a means of increasing the efficiency of RNA translation.
[00244] These findings confirm the results of Example 12, demonstrating that ψmRNA is more stable than unmodified RNA.
[00245] Further immunogenicity of ψ-mRNA was less than unmodified RNA, as described herein above (Figure 7 and Figure 12C, right panel).
[00246] To summarize Examples 13-14, the 3 advantages of ψmRNA compared with conventional mRNA (enhanced translation, increased stability and reduced immunogenicity) observed in vitro are also observed in vivo.
305. Claims 1 and 2 of UPenn are:
1. A messenger RNA comprising a pseudouridine residue.
2. The messenger RNA of claim 1, further comprising a poly-A tail.
306.
It is convenient to set out here the structures of the 5 molecules listed in [0056] (taken from Dr Enright's evidence):
307. Pfizer/BioNTech's case of anticipation was set out in an extremely useful chart with its closing submissions. There were three routes, as I will describe further below.
308. The first two are based on Example 31 of UPenn and the third is not, but rather is based on [0056] (or claim 1 of UPenn). The second is a relatively minor variation on the first.
309. I will refer to them as Route 1, Route 2, and Route 3.
310. There is a little bit of an overlap in terms of "pointers" relied on, but Route 3 is really very distinct from Routes 1 and 2.
311. To succeed fully, Pfizer/BioNTech needs to show that claim 3 and claim 5 of EP949 are both anticipated, with claim 5 adding the additional requirement of a polyA tail. Pfizer/BioNTech argued that the polyA tail followed inexorably from Route 1; to the extent that any additional selection was needed it sought to justify that within Route 2 and Route 3.
312. I remind myself that claim 3 requires an mRNA, that m1Ψ has to be used as a modified nucleotide, and that 100% of the uracils have to be replaced.
313. Route 1 involves starting with Example 31, with its reference back (at [0290]) to use of the methods described in Examples 2 and 7, and the list of 96 nucleosides in [00291] including m1Ψ.
314. Pfizer/BioNTech's Route 1 argument is that m1Ψ is individually disclosed in [00291] and that [00290] individually discloses the use of Example 2 and of Example 7 with each of the 96. Going back to Example 2, Pfizer/BioNTech say that 5 RNA sequences are disclosed, of which RNA-1866 is an mRNA and has a polyA tail. It should be noted that none of the other 4 has a polyA tail and that RNA-713 at [00191] is not an mRNA (but an anti-sense strand: see [00187]).
315. It is common ground that to the extent that the skilled person undertook Example 2, replacement with the modified nucleoside would necessarily be 100% given the method described at [00193].
316. It is convenient also at this point to mention that the use of m1Ψ necessarily means that uracil will be replaced (not just in Example 2, but generally); in the Netherlands (and possibly the EPO) Moderna has argued that the choice of uracil to be replaced is an additional choice but this is wrong or at least inaccurate and although there was a faint hint of it in Moderna's written submissions it was not supported in oral argument.
317. Pfizer/BioNTech said that it was irrelevant that more than one canonical nucleoside can be replaced because the claim only positively requires the replacement of uracil. I agree with this and Moderna did not make anything of the point. Pfizer/BioNTech also said that it did not matter that Example 7 involves a lesser degree of replacement than 100% because (a) it is taught to be done on top of Example 2 and not as an alternative and (b) Example 7 involves making a 100% replacement as well as lower percentages, as can be seen for m6A, Ψ and m5C in Figure 5.
318. As I have mentioned, Route 2 is a variant on Route 1 and comes into play in case I should conclude that a selection from the RNAs in Example 2 is needed to get to claim 3 (as opposed to each of them being disclosed in combination with each of the 96 options from [0291]). The arguments as to claim 5 overlap with those on claim 3 here.
319. Pfizer/BioNTech's argument on Route 2 was that there were pointers to help it:
i) mRNAs are translated;
ii) the skilled person would understand UPenn to be about modified RNAs giving reduced immunogenicity and enhanced translation;
iii) Example 31 describes testing the effect of nucleoside modifications on efficiency of translation;
iv) Four out of five of the RNAs in Example 2 encode proteins and are mRNAs; and
v) Claim 1 of UPenn refers to "A messenger RNA comprising a pseudouridine residue".
320. Pfizer/BioNTech said these were "pointers" towards choosing an mRNA (all that is needed for claim 3) and additionally, for claim 5, towards choosing RNA-1866 which has the polyA tail. Additionally, on claim 5, Pfizer/BioNTech relied as pointers on:
i) Claim 2 of UPenn;
ii) The fact that RNA-1866 is one of the two RNAs in Example 2 that encodes a luciferase (the other being RNA-1571), and almost all of UPenn's experiments are directed to luciferase;
iii) RNA-1866 is the only RNA in Example 2 that has a polyA tail which would be preferred for protein production, stability and enhanced translation; and
iv) Prof Rosenecker had said that Example 12 showed that the effect of the cap and tail together were greater than the effect of Ψ itself.
321. Moderna made the following main points on Routes 1 and 2.
i) That the list in [00291] is long and "copy-pasted".
ii) That Example 31, written in the present tense, would be understood to be prospective things that could be considered, rather than things that had been done. It relied on evidence both from Dr Enright and Prof Rosenecker that the Example was a "suggestion" that one "could" go and "try" things, "potential modifications". Usually, evidence from experts on the meaning of a disclosure would be regarded as inadmissible because the question is one for the court, but here I think technical understanding is useful and necessary to appreciate the real gist of what is being proposed. But in any event, I would reach the same conclusions as the witnesses.
iii) That Example 31 is open-ended. I agree with this. Its whole tone is open ended, although the most concrete textual example is "include e.g." at line 10 on page 74.
iv) That the total number of things proposed, multiplying the 96 possibilities in [00291] by the number of RNAs and methods in Example 2 and Example 7, was enormous and would take a very long time. Dr Enright accepted that the skilled person would not think they were being told to do all the possibilities, and again I think expert input is admissible although I would reach the same conclusion myself without it.
v) mRNA is not inevitable given the presence of RNA-713 (this is factually correct).
vi) RNA-497 (which is an mRNA) was also not in fact tested in Example 2 given that it is shorter than the minimum length indicated at line 20 on page 48 ("lengths between 0.7-1.9kb"). I find that this is also correct.
vii) While translation efficiency is one of the things for testing proposed in [00290] it is not the only one and anyway a polyA tail is not necessary for translation, even if it may enhance it. I agree with this, too.
viii) Although the Examples of UPenn do indeed nearly all concern luciferase, most of those that do so used RNA-1571 which does not have a polyA tail. This is factually correct, as Prof Rosenecker's evidence showed in a detailed annexe.
ix) [0051] of UPenn describes the polyA tail as optional and it is not permitted to make a pointer out of an option by adding CGK. It is true that [0051] says this, but the point is undermined considerably by claim 2.
322. In my view Moderna has much the better of these arguments and there is no anticipation by Route 1 or Route 2.
323. Although I accept that it is not necessary for anticipation for a prior art document to say that something has actually been done (so that I reject any argument by Moderna that Example 31's being prospective is a reason in itself to reject anticipation), Example 31 is extremely tentative and open ended. The extent of what it is proposing and the reasons for doing it are both woolly (see Jacob LJ in Dr Reddy's at [31]).
324. I also consider that all that Example 31 is saying in relation to Examples 2 and 7, at least with any clarity, is that they provide methods that may be used. I do not see any teaching in Example 31 to go back and redo Example 2 or Example 7 with the exact RNA sequences disclosed there.
325. Of course, I agree that m1Ψ is present in black and white in the middle of the list in [00291] but that does not of itself make it an individualised disclosure, as is clear from Dr Reddy's. Similarly, the fact that the options are given in the form of a list does not make it so. I have concluded above that there is no precise numerical limit but with that said, I do not find there anything shocking in the rejection of one from among 96 as not being individualised, and in any event the list is in fact open ended given "include, e.g.".
326. Even if I were to regard m1Ψ as individually disclosed in Example 31, I would not have accepted that there was no need for any selection from lists. m1Ψ is one of a long list with no statement of preference for it (Pfizer/BioNTech's obviousness arguments cannot be used for this) and if it is individually disclosed it is only in this sense and in this context. To get to anticipation it would be necessary to take m1Ψ and use it with an mRNA of Example 2, specifically RNA-1866 (for claim 5) or RNA-1571 or RNA-730. Since Pfizer/BioNTech need to knock out claim 5, the key question is over RNA-1866.
327. In my view this is a selection from lists, one of uncertain extent but at least 96 long, and one of five (assuming, contrary to my analysis above, that one reads Example 31 as directing attention back to the specific sequences in Example 2).
328. As well as the length and nature of the lists one also has to consider "pointers". Although there is something for Pfizer/BioNTech to work with in that sense, its best case being that polyA is called out in claim 2 and that mRNAs would be especially useful for some of the purposes pointed out in Example 31, it is far from all one way because of Moderna's points that UPenn itself mainly works with a non-polyA tail sequence, and that one sequence in Example 2 is not an mRNA at all. It should be recalled that "pointers" are things which flag up something that sets aside the relevant element of the list from the others and that the standard is clear and unmistakable disclosure. Pfizer/BioNTech's points do not rise to that standard. The whole exercise smacks of arguments for obviousness and not anticipation.
329. I would also point out that even if there is a "pointer" within Example 2 to RNA-1866, it is much weaker to select based on a preference from one list and to select arbitrarily (i.e. not based on preference or "pointer") from another list, which is what Pfizer/BioNTech is doing, given the lack of any preference or "pointer" in relation to m1Ψ.
330. Route 3 depends on paragraphs [004], [0056] and [0074] of UPenn. An alternative to [004] is claim 1.
331. Pfizer/BioNTech argue that either [004] or claim 1 points to, and discloses, "a pseudouridine". It then relies on [0056] as defining what is meant by "pseudouridine", which is Ψ itself, plus the four other named possibilities, plus "any other pseudouridine known in the art". Then, the 100% replacement is said by Pfizer/BioNTech to be disclosed in [0074] in a list which includes 100% as well as numerous other possibilities.
332. Pfizer/BioNTech said that there are "pointers" to 100%, to which I will return in a moment.
333. For claim 5 it relies on claim 2 and the statement in [0051] that having or not having a polyA tail are each options. It fortifies this by reliance on the same polyA tail "pointers" as under Route 1 and Route 2.
334. Although positioned as Pfizer/BioNTech's secondary case, I found Route 3 simpler and rather more persuasive than Route 1 (or Route 2), but I nonetheless reject it.
335. I can accept that claim 1 or [004] is at least a legitimate starting point, but both routes lead to [0056]. [0056] is an odd creature because it is a definition section and not an expression of any technical preference for the listed nucleosides (a point I have considered in relation to obviousness, too). It is also open-ended because of the reference to any other pseudouridine known in the art. Although the skilled person could look into this and discover from the RNAMD that there were no other known naturally-occurring pseudouridines, this is a process of research and deduction plugging in aspects of the CGK, not part of the disclosure of UPenn. Anyway, the skilled person would then know that there were no other artificial pseudouridines in the RNAMD, but not that there were no others in the world, and would also think that the author might well be referring to ones that might be found in future.
336. In any event, m1Ψ is not said to be preferred.
337. As to pointers to 100%, it is true that Example 2 gives a method for achieving 100% replacement but that does not necessarily make it preferred in terms of what it could actually do (a lesser percentage might be just as good); I do accept that achieving 100% would be seen as easier to do and more reliably than the other possibilities, but the same applies. Pfizer/BioNTech argued that 100% would be preferred because of the teaching that greater modification would imply greater immunosuppression, but that is not the same as saying that 100% would be necessary, not least because the whole point of Example 7 is to assess how much replacement would have what effect.
338. I therefore think that these pointers are not strong, are not explicitly disclosed but require analysis, and are not directly linked to the list at [0074].
339. But even if I assumed, contrary to those conclusions, that there was a strong pointer in favour of 100% replacement, Pfizer/BioNTech's case suffers badly from the absence of any pointer to the combination of m1Ψ and 100% replacement and that, together with the fact that its case required combining something preferred (100% replacement, on this assumption) with something in an unclear and apparently open-ended list (m1Ψ), leads me to reject Route 3 as well.
340. EP949 was opposed in the EPO by nine parties, including BioNTech and Pfizer.
341. I was provided with a preliminary opinion which was issued by the Opposition Division ("OD") on 8 December 2023. Claims 1 and 3 were in issue and both of the prior art citations relied on by Pfizer/BioNTech at this trial were considered by the OD.
342. The OD's preliminary opinion was that EP949 was invalid for added matter and lack of inventive step over UPenn, but that the novelty attack over UPenn was not convincing.
343. During the trial before me the oral proceedings took place in the OD and EP949 was upheld in amended form, in which there was only one claim, the same as claim 3 before me.
344. The OD's preliminary opinion on the novelty attack was in two parts. First, in relation to the attack based on [004] and [0056] (see 5.7.2):
i) The OD found that the features require a selection to be made from equally preferred alternatives;
ii) The OD found no pointer within UPenn which would cause the skilled person to combine the embodiments in the way required. The combination of features would result in a "specific subset of meanings which was not originally disclosed in D11 [UPenn]".
345. The opponents also argued that Example 31 of UPenn anticipated the subject because it incorporated the methods used in Examples 2 and 7, and in reliance on Example 2 disclosing the feature of 100% replacement of a given nucleotide for the modified one. The opponents argued that this was an implicit disclosure of the preparation of an mRNA wherein 100% of uridines are replaced by m1Ψ.
346. The OD dealt with this submission on the following basis (see 5.7.2):
The Opponents' reasoning requires a combination of examples 2, 7 and 31, with no disclosure or even suggestion pointing towards the 100% replacement of uridine with m1Ψ as being preferred over the whole range of possible replacements. In this context it is further noticed that example 31 provides a list (cf. [00291]) of possible modified nucleotides but does not provide any experimental data pertaining to the synthesis, let alone the characterisation of the (allegedly) synthesised mRNAs encompassing any of the modified nucleotides therein listed. Thus, on the one hand, the said list cannot be considered as disclosing a list of synthesised mRNA derivatives. On the other hand, there are no indications that the reference to m1Ψ in the said list must unambiguously be considered as referring to an mRNA residue resulting from the 100% replacement of the uridine moiety.
Example 31 may thus at best be seen as suggesting that the mRNA as defined in claim 3 of the opposed patent may have been synthesised. However, novelty cannot be a matter of probability.
347. The OD's reasons in the preliminary opinion are consistent with mine, albeit briefly stated and provisional. I do not know whether the OD's analysis at the opposition hearing was the same, or different, however, because its reasons are not available yet. Pfizer/BioNTech pointed out that it is possible that the OD relied on a bad argument that I would not have accepted (such as the choice of uracil being an independent one from the choice of Ψ). This is possible although, I think, unlikely. In the round, I am glad that I have reached a consistent conclusion and think it is likely that my reasoning coincides fairly closely with the OD, although I am confident I would have arrived at the same result in any event.
348. An anticipation attack over UPenn was also advanced in parallel litigation in the Netherlands.
349. By a decision of 6 December 2023 in case C/09/643000/HA ZA 23-169, the District Court of the Hague found EP949 to be invalid for lack of novelty over UPenn and I was provided with the judgment ("the Dutch Judgment") dated 6 December 2023.
350. The District Court of the Hague described the subject matter of UPenn and EP949 as follows (see paragraph 4.16):
WO 708, like EP 949, pertains to a method to manufacture mRNA in which nucleotides have been modified. Both patents mention the purpose of the invention disclosed therein to manufacture mRNA that - thanks to modification of certain nucleotides - has improved properties in terms of protein production (translation capacity) and suppressing the innate immune response (immunogenicity) (cf. [0038] of WO 708 and [0008] of EP 949). Both in WO 708 and in EP 949, this is essentially resolved by replacing (uracil) nucleotides with nucleotides with a (uracil) variant (insofar as relevant here: Psi and/or m1Psi). Moderna identifies two characteristics in claim 1 that would be missing in WO 708: (1) m1Psi and (2) 100% replacement of the uracil nucleotides.
351. At paragraphs 4.17 to 4.19 of the Dutch Judgment, the Court considered [0056], [0074], Example 2, Example 7 and Example 31 of UPenn. Moderna argued that a choice from three lists must be made to get to the claimed subject matter (see paragraph 4.20):
i) The choice from the list of four nucleotides;
ii) The choice from the list of different mRNA modifications; and
iii) The choice from the list of 0-100% substitution.
352. Moderna's submission was rejected by the Court. The Court agreed with Pfizer/BioNTech's submission that the lists were dependent on each other and therefore the case law relating to two or more independent lists of different components was not applicable. The Court said that it was inherent in the choice of m1Ψ that a replacement at the site of uracil takes place (I agree with this), and that a technical advantage for choosing 100% modification can be found in UPenn at [00218] (see paragraph 4.20).
353. The Court went on to consider Moderna's submission that Example 31 of UPenn does not directly disclose the claimed subject matter:
4.23 Moderna should be credited that WO 708 does not explicitly describe the experiment in which mRNA is made with an m1Psi modification, in which 100% of the uracil nucleotides are replaced by m1Psi. However, the court agrees with the defendants that the average practitioner, using general professional knowledge, does implicitly infer all these features of conclusions 1 and 3 of EP 949 in a direct and unambiguous manner from WO 708, more specifically from par. [0056], Example 31, which refers to Examples 2 and 7, and par. [0074]. Although the list in Example 31 is a standard list that includes many modifications, this does not detract from the fact that the ml Psi modification is disclosed individually. It is not an undue burden for the average practitioner to understand that a modification from the list such as ml Psi must be built into the mRNA in the manner described in Example 2 and 7, thus replacement of 100% of the nucleotides. Inherent to the choice of ml Psi (a pseudo-uridine) is that (all) uracil nucleotides are replaced with this. It is also sufficiently clear that the average practitioner can also make mRNA modified without undue burden.
4.24. Moderna's argument that the standard list that is disclosed in paragraph [00291] also includes modifications that are not/do not appear to be workable, does not change the fact that the m1Psi modification is individualised with 100% replacement therein. Moderna did not provide any arguments as to why the average practitioner had reason to assume that the list shown in paragraph [00291] of WO 708 was entirely speculative on the priority date of EP 949. It only submitted that it was later found that some of the modifications mentioned therein did not work. In addition, WO 708 describes in paragraph [0056] a preference for four Psi derivatives, including m1Psi. The average practitioner - especially bearing in mind the structural similarity to successful modifications as mentioned above in ground for legal consideration 4.21 - has no reason to assume that m1Psi will not work and then only has to test this modification; it is not necessary that this person tests the entire list of modifications. The court also rejects Moderna's argument that novelty requires that the practitioner should know when seeing the list (in Example 31 and paragraph [0056]) respectively) which modifications would and would not work. Contrary to what it argues, there is indeed an instruction in WO 708 how the practitioner can invent this without undue burden, namely by carrying out the tests performed therein (of Example 2 and 7).
Fn: cf. Boards of Appeal. 14 March 2019, T 464/15, ECLI:EP:BA:2019:T046415.20190314 (Targeted DNA insertion/Bayer Cropscience), paragraphs 23-25 and Boards of Appeal 1 February 2023. T 809/16, ECLI:EP:BA:2023:T080916.20230201 (Opioid dosage farms for reducing adverse effects associated with alcohol-induced dose dumping/ALZA) paragraphs 3.7-3.8.7).
4.25. This leads to the conclusion that, in the court's opinion in WO 708, the average practitioner on the priority date finds a method of making an mRNA molecule in which 100% of the uridines have been replaced by m1Psi. A lecture with a "mind willing to understand" and using the general expertise, does not allow any other conclusion. This means that claims 1 and 3 of EP 949 are fully disclosed in WO 708, so that EP 949 is not new according to those claims.
354. The Court also considered that its decision was strengthened by the "unhindered granting of two patents applied for based on [UPenn] (EP 3 611 266 and EP 2 578 685), which claim the use of (Psi or) m1Psi in modified RNA" (paragraph 4.27).
355. The fact that an experienced and respected tribunal which also applies the EPC has reached the opposite conclusion to me naturally causes me to stop and check my reasoning and my conclusion. I would of course like to understand the potential reasons for the difference. I think there are several reasons. One is that the Court relied, in passages not quoted above, on evidence that was not before me; evidence is sometimes, although relatively rarely, admissible and relevant to novelty but in any case this is a difference between those proceedings and these. An example of this is that the Court found there was no evidence that [0291] in UPenn was speculative, but Dr Enright agreed that it was. A second is that the arguments were different, for example I note that the Court relied on implicit disclosure and undue burden, which is not how Pfizer/BioNTech put matters before me. A third is that the Court found some factors to be persuasive which, as a matter of evaluation I do not. For example, I do not think that the grant of patents based on UPenn claiming the use of m1Ψ in mRNA carries any weight as such, without knowing what the arguments and the reasons for any decision were.
356. I also note that the Court did not go on to decide obviousness over UPenn. Of course, it did not need to having upheld the anticipation attack. It is however possible that the Court would have found the Patent obvious based on the evidence that it heard (different from that which I heard) and indeed the points advanced by Moderna why the Court's decision on anticipation was wrong would not run, or at least not nearly so well, in relation to obviousness. So in the end it is possible that the real and underlying difference between the District Court of the Hague's decision and mine is in relation to obviousness, and explicable by different evidence.
357. In any case, my conclusion on anticipation was bound to be inconsistent with either the EPO or the District Court of the Hague and I prefer the former in circumstances where, on the materials available, it considered the same arguments that I did and where its decision was less dependent on (different) expert evidence.
358. The central dispute between the parties was whether the skilled person would carry forward investigations into Ψ-modified mRNA for use in therapy having been encouraged by the results disclosed in UPenn or Karikó 2008 (as Moderna contended), or whether the skilled person would instead be encouraged to investigate further mRNA modifications (other than Ψ-modified mRNA) upon reading UPenn or Karikó 2008 and consulting the RNAMD (as contended by Pfizer/BioNTech), and if they did the latter, what work they would then take forward without invention.
359. I will deal with obviousness over UPenn first and then consider whether any different conclusion would be justified over Karikó 2008. Neither side argued with any force that it should, but there are a number of points of difference that were raised.
360. I summarise Pfizer/BioNTech's case as follows:
i) The data on Ψ in UPenn were very promising and of real interest.
ii) The skilled person would decide to explore other nucleoside modifications.
iii) The ones of interest would be the "Ψ-like" ones.
iv) It would be possible to make m1Ψ if that were selected.
v) If the skilled person made m1Ψ the most obvious thing to do would be to modify 100% of the uracils and to include a polyA tail.
vi) Carrying out the necessary translation and transfection experiments as in UPenn in vitro would take a couple of months.
361. Point i) was not in dispute. Indeed Moderna made a virtue of it by contending that Ψ was so good that the skilled team would put all their efforts into deploying it in a practical way; in the context of Prof Rosenecker's expertise that would mean moving towards using it in transcript therapy, or for other applications, for them.
362. I did not detect that point iv) was materially in dispute. The same applies to point v). As to point vi), there was a dispute but not one that independently affects my decision either way: I think that with a sufficiently strong and appropriate motivation and sufficiently clear way forward, the effort involved would not in general deter the skilled person. The effort involved in a shotgun approach, working blind, would be much greater, though. I consider the nature of the motivation and way forward in detail below.
363. Thus the main disputes were on the effect of point i) - would the success with Ψ lead to progressing it alone or to making other modifications - and on point ii) and point iii). I do not overlook that the totality of all the steps has to be considered, so the fact that some steps were not disputed individually does not mean that they fall completely out of account, but I think there is really so little in steps iv) and v) that no more need be said about them.
364. Although Pfizer/BioNTech's case is thus presented as involving "just two decisions" - steps ii) and iii) - there is in my view a lot of thinking and decisions packed into each of them.
365. First, however, I will consider step i) a little more. Moderna's position was that because transcript therapy specifically had been plagued with problems in the period up to the EP949 Priority Date, the natural thing to do would be to try Ψ in that context. In other words, the point was based on Prof Rosenecker's perspective in transcript therapy and I do not know if it would apply to other fields of application of mRNA as discussed in the section on the skilled person.
366. This undermines the force of the point in Moderna's favour but it does not entirely negate it. I think it remains of some significance because of the fact, which I come on to in relation to points ii) and iii), that the skilled person would not understand the reason for Ψ's good results. That would naturally lead them to think that they should first see if Ψ was of practical utility in their own field before branching out.
367. Additionally, this point was said by Pfizer/BioNTech to be lacking weight because it is a Brugger v Medicaid point, and that it being obvious to try Ψ out does not make it any less obvious to try other modified nucleosides. I do not think Brugger means that this sort of point cannot have weight, it just means that the presence of a particularly attractive way forward does not necessarily make other obvious things non-obvious.
368. A further argument of Moderna's, related to this, is that the skilled person might carry on with Ψ and, instead of modifying the nucleoside, try to improve the cap and polyA tails. Pfizer/BioNTech said that it was illogical for Prof Rosenecker to say that the skilled person might do this while denying that they would change the nucleoside modification but I disagree, because there was some understanding about how the former worked and none about the latter. So this is another available avenue of work from UPenn that I think would be more logical than changing the nucleoside modification and is another factor.
369. So I think this aspect of point i) and the options to stick with Ψ and/or change the cap and tail, have some modest heft for Moderna as part of the overall picture, but I would have reached the same ultimate conclusion even if I disregarded it altogether.
370. Point i) also naturally goes along with the fact that the skilled person would appreciate that the scientists behind UPenn were of the highest quality and eminence in the field. Moderna did not dispute this and I take it into account, but it does not have much importance because it was not Moderna's case that the science in UPenn would be seen as poor - quite the opposite - but just that it does not point to m1Ψ by obvious means or reasoning.
371. I move on to points ii) and iii). Although I have split them out above, they are closely interrelated, since, for example, it is the focus which Pfizer/BioNTech says would be put on [0056] that among other factors would motivate the skilled person to make other nucleosides in the first place.
372. The necessary starting point is the disclosure of UPenn itself.
373. Pfizer/BioNTech said that UPenn contained three lists of modified nucleosides, in [0056], [0069] and [0070].
374. I have considered [0056] in the context of anticipation above and in the context of assessing the hindsight point Moderna made against Dr Enright.
375. [0069] is a list of the modifications tested in UPenn, and to the extent it matters I agree with Pfizer/BioNTech that the skilled person would realise that.
376. [0070] is a much longer list of 92 possibilities. Pfizer/BioNTech said that this was derived from those naturally occurring modifications listed on the RNAMD, less the ones already called out in [0056] and [0069]. I agree that the skilled person could check this, but I do not agree that it would occur to them spontaneously, and I do not think Dr Enright said it would.
377. In addition to these three lists, there is of course [00291] within Example 31, which contains 96 possibilities: it has the [0056] list added to the [0070] list, less the ones already tested. I have discussed [00291] above in connection with anticipation and pointed out that the skilled person would think it was prophetic; it does not mean that the modifications listed had in fact been tested, only that they could be. The object of that testing would be to assess their immunogenicity and translation (see [00290]). I agree with Pfizer/BioNTech that the skilled person would think that translation was of importance because the advance of UPenn over Karikó 2005 was in that area.
378. Pfizer/BioNTech's case was very heavily dependent on [0056]; for example Prof Rosenecker said that Karikó 2008 provided no direction for the skilled person and Pfizer/BioNTech's written closing argued that that "cannot apply to UPenn given [0056]". [0056] was put to Prof Rosenecker forcefully during his cross-examination and he maintained his position that it was baffling. See for example T4/51914 to T2/52112:
Q. And the skilled person would understand that the authors of the UPenn document were suggesting that the next batch of nucleotides to try were those listed in paragraph [0056]?
A. They say that?
Q. No, I am suggesting that the skilled reader of this document, they would see two groups of nucleotides that are listed in small groups.
A. Yes.
Q. Paragraph [0056] and paragraph [0069].
A. Yes, but ----
Q. Just let me put the question again, professor.
A. Yes, yes.
Q. They would understand that the nucleotides in paragraph [0069] had been tested?
A. They had been tested, right, yes.
Q. And they would understand that the authors of this document were inviting the reader to test the molecules in paragraph [0056] next, because those had been ----
A. I do not agree. I do not agree.
Q. Why else do you think that Karikó and Weissman have listed the pseudouridines in paragraph [0056]?
A. I do not know.
Q. I suggest to you the skilled person would not just say "I do not know" and stop thinking, they would understand the reason that they had been listed was because Karikó and Weissman were suggesting that these were also useful modified nucleotides?
A. But why not testing the ones named in [0070]? There is another list in paragraph [0070] of, I do not know, 90 or whatever it is. I do not see this as an invitation to test now all the compounds listed in paragraph [0056]. The skilled person would not understand why the compounds in paragraph [0056] are listed.
Q. Can I suggest to you that the skilled person would see that they are most like pseudouridine, of the molecules listed in the document, and ----
A. They are -- excuse me.
Q. Do you agree with that, they are most like pseudouridine?
A. They are named pseudouridines.
Q. And they are the molecules most similar in structure to pseudouridine, putting aside perhaps m5D for now, but apart from m5D, they are clearly the molecules most similar to pseudouridines listed in the document?
A. They are pseudouridines, yes.
Q. They would therefore be seen as the most promising nucleotides to try to see if the results with pseudouridine could be equalled or bettered?
A. Why? There is no indication to test those in [0056].
379. I do not think that [0056] will bear the weight that Pfizer/BioNTech sought to put on it:
i) It does not naturally read as a technical teaching that the listed nucleotides are preferred or particularly beneficial. It is a definition not a scientific statement.
ii) The presence of m5D makes it unclear what thinking had gone into the list. I cover this above in relation to Dr Enright's hindsight.
iii) It is not a concrete list of 5 possibilities because it also extends to any other pseudouridine known in the art. I agree that if the skilled person went to the RNAMD they would find there were no other natural pseudouridines, but they would not know the position about artificial ones.
iv) There are multiple other lists in the document.
v) The list in relation to which there is a positive teaching to look for other, better, nucleosides is [00291] which is a technical teaching, albeit a broad and prophetic one, accompanied with methods and so on.
380. UPenn has to be read as a whole in this regard, and it is certainly not legitimate, as Pfizer/BioNTech sought to do at one point, to give primacy to [0056] just because it comes first.
381. An additional tension in Pfizer/BioNTech's case on this point is that it seemed to involve the unspoken assumption that there would in fact be modifications better than, or as good as, Ψ among any that the skilled person decided to test. But Dr Enright accepted that he or she would not know even that.
382. A further and closely related point which I think is important and powerfully in Moderna's favour is that UPenn does not say why Ψ had worked so well. Nor did Pfizer/BioNTech have any real case that it was possible to work out why (it did submit that in relation to Karikó 2008 there was a teaching that the improvement arose from stability, but that does not help because it did not say what about Ψ provided the stability, "no mechanistic rationale" as Pfizer/BioNTech put it). The skilled person's reading of [0056] and e.g. [00291] would be informed by this. So to this extent the choice of other modified nucleosides to try would be made "blind" and there could not be any inference that the choice in [0056] was made on a concrete basis of understanding the mechanisms at work. Indeed, Pfizer/BioNTech's case was in large part, in reality, that the skilled person would set about making modifications in order to understand why Ψ had produced such good results. In another passage of his evidence heavily relied on by Pfizer/BioNTech, Prof Rosenecker agreed that that could be done, but it is important that it went no further than could.
383. The next aspect of Pfizer/BioNTech's case to consider in connection with points ii) and iii) is the RNAMD. Pfizer/BioNTech's case, supported by Dr Enright's evidence, is that the skilled person would cross check the [0056] list against the RNAMD. Assuming that in Pfizer/BioNTech's favour, as I think is reasonable on the evidence, doing so would confirm that there were no other naturally occurring pseudouridines (as I have mentioned above). But I think it would also lead the skilled person to consider what other information was available about the 5 modified nucleosides from [0056]. In relation to m1Ψ there is a sub-issue, which I regard as an important one, about a review article called Charette & Gray, to which I will digress in a moment.
384. I also note that Dr Enright was not consistent in his approach to the list in [0056]: he both suggested that the skilled person would prioritise them, and that the skilled person would make and test them all. This inconsistency arose, I think, from the weakness of the skilled person's ability to prioritise in a way in which he or she could have confidence, and it undermines the case for obviousness all the more.
385. Charette & Gray is a review article entitled "Pseudouridine in RNA: What, Where, How and Why", Life 2000; 49: 341-351.
386. Moderna submitted that if Pfizer/BioNTech's case were to be accepted up to the point of the skilled person looking in the RNAMD, he or she would then look at Charette & Gray, because it is referred to in the Comment section for pseudouridine in the following terms: "Leading references to the pseudouridine literature can be found in a recent review [18]". Prof Rosenecker said a reason for this course was because the skilled person would have no understanding of why pseudouridine outperformed uridine; a more general reason would be a desire to get an overview of pseudouridine's characteristics.
387. Pfizer/BioNTech did not directly contradict this reasoning and I accept it. Dr Enright was doubtful whether the skilled person would read all the references given for pseudouridine in the RNAMD, but might want to look at Charette & Gray to understand pseudouridine's better performance.
388. Pfizer/BioNTech also questioned why Prof Rosenecker had said that the skilled person would read Charette & Gray to the exclusion of all the other references in the RNAMD given for pseudouridine.
389. I do not accept this sniping by Pfizer/BioNTech. I think it is plain that if the skilled person went to the RNAMD for pseudouridine, as Pfizer/BioNTech argues they would, and read it carefully, as it must be assumed they would, then Charette & Gray would stand out as a key reference for general information and the skilled person would read it. The other references would look of much less use or interest.
390. I turn to what Charette & Gray would teach the skilled addressee. The relevant parts are the following from the Summary:
Through its unique ability to coordinate a structural water molecule via its free N1-H, Ψ exerts a subtle but significant "rigidifying" influence on the nearby sugar-phosphate backbone and also enhances base stacking. These effects may underlie the biological role of most (but perhaps not all) of the Ψ residues in RNA.
391. And the following paragraphs of the section "Structure and Physicochemical Properties of Pseudouridine":
...The special properties of Ψ relative to U are largely attributable to the former's additional hydrogen bonding capabilities. In the anti conformation, Ψ in RNA provides the appropriate geometry and distance for coordination of a water molecule between its N1-H and the 5' phosphates of both Ψ and the preceding residue. In cases where such a water bridge has been inferred, this restricts base conformation and mobility of the backbone 5' to the site of pseudouridylation, regardless of sequence or structure (single- or double-strand) context (see Fig. 2).
Pseudouridylation has the additional effect of enhancing local RNA stacking in both single-stranded and duplex regions by favoring a 3'-endo conformation of the ribose, which restricts the base moiety to an axial anti conformation (see Fig. 2). Here again, a structured water molecule is implicated in the mechanism of this stabilization, which involves replacement of a weak C5-H...OW interaction in U by a stronger N1-H...OW hydrogen bond in Ψ... In fact, improved base stacking has been proposed to be the most important contribution of Ψ to the stabilization of RNA structure.
392. I have omitted the references from the above, but there are a quite number of them, and relevant to the arguments at trial are the references at the end of the second quote above, which are "(5, 11, 12, 14, 15)". Reference 12 is a paper by Yarian, which I address further below.
393. Two theories are relevant here and were the subject of evidence and submissions before me. One is hydrogen bonding, which Charette & Gray mention first. If the skilled person accepted that, it would tend to point away from m1Ψ. The other theory is base stacking, in connection with which Pfizer/BioNTech relied on Yarian et al, "Structural and functional roles of the N1- and N3-protons of Ψ at tRNA's position 39", Nucleic Acid Research 1999; 27(17): 3543-3549 ("Yarian").
394. Yarian comments on both the theories, and asserts that increased stability comes from base stacking and not hydrogen bonding, as summarised in the following:
By incorporating the methylated derivatives of Ψ, we were able to block the ability of the Ψ N1 and/or N3 positions to hydrogen bond and examine if the stabilizing effects of Ψ were still present. When the proton from the N1 position of Ψ was replaced with a methyl group in the tRNAPhe ASL construct, the ASL actually exhibited an increased stability resulting in an increase of 2.0oC in Tm as compared to the ASL-Ψ39 (Table 1). In comparison, when the isosteric proton in uridine at position 5 was replaced with a methyl, the Tm increased by only 0.3oC compared to the unmodified ASL. These results suggest that the increased stability from incorporating Ψ39 resides in the nucleoside's ability to stack with a more thermodynamically favored arrangement and not from additional hydrogen bonding. Methylation of the N1 position improved pseudouridine's ability to stack in comparison to uridine.
395. Pfizer/BioNTech relied on Yarian in two main ways: first, that it says that the correct explanation is base stacking and not hydrogen bonding, and second that it actually contains data about methylation at the N1 position, which, Pfizer/BioNTech said, showed it was more stable than Ψ with better base stacking.
396. Moderna responded that Charette & Gray post-dates Yarian and is a review article, in which the authors had maintained the hydrogen bonding theory.
397. Pfizer/BioNTech also said that Charette & Gray's theories only applied to specific positions in tRNAs and possibly rRNAs.
398. The above matters were explored in some detail with Prof Rosenecker and Dr Enright.
399. A feature of the written and oral evidence on this complex topic which I think is important, is that Charette & Gray and what Moderna would be saying about it were flagged very clearly in Prof Rosenecker's second report. By contrast, Yarian only came into the case in the cross-examination bundles, and Dr Enright did not give written evidence about either. It is quite obvious from the way the relevant paragraphs of Charette & Gray are written, from the number of references there, and from their titles, that Charette & Gray had reviewed more literature than Yarian. So it is all the more plausible that the reader of Charette & Gray would think that the authors had concluded that the hydrogen bond theory was sensible, and any weight I could attach to Yarian is substantially undermined by the sense that Pfizer/BioNTech's reliance on it is probably selective, and by the lack of evidence in chief from Dr Enright on it. Counsel for Pfizer/BioNTech said that Moderna should have seen that Yarian was in the cross-examination bundle and looked into other references in Charette & Gray, but I reject that as unrealistic and unfair. Moderna had set its stall out properly and in good time and the onus was on Pfizer/BioNTech to respond in an appropriately full and timely way.
400. Although the picture is a complex one, in my view the skilled person would, on balance, be more likely to go to Charette & Gray on the basis that it is the review article commended by the RNAMD, read its summary, and stop there, taking on board that it endorsed the hydrogen bonding theory which, if applied to m1Ψ, would tend to suggest reduced stability. I do accept on the basis of the oral evidence that the skilled person would not think this was a theory that was supported by strong evidence, or the only theory, and they would take on board that its application to mRNA was a matter of uncertainty. Nonetheless, in a situation where the skilled person would otherwise have no basis for working out why pseudouridine achieved what it did, it would be the best information and analysis available. I reject the notion that the skilled person would dig deeper than Charette & Gray, but if they did then they would not go exclusively to Yarian, and Pfizer/BioNTech, whose burden I think it was, for reasons mentioned above, did not provide any evidence of what a thorough review of the references in the relevant part of Charette & Gray would yield.
401. I also agree with Moderna that even if the skilled person did attempt to take on board Yarian, the overall picture would be very confusing and provide no clear pointer.
402. Pfizer/BioNTech's last throw of the dice on this point was to suggest that if the skilled person took on board Charette & Gray's theoretical analysis, or had also read Yarian and noted the disagreement, and therefore had doubts about m1Ψ, the way to resolve the uncertainty would be to conduct a test. I think this was very speculative and Prof Rosenecker conceded no more than that it would be possible to test.
403. Returning to Pfizer/BioNTech's reliance on [0056], Dr Enright gave evidence about how the skilled person would think through and rank the list of 5 modifications in [0056] were they to get that far (although as I have mentioned above he was not wholly consistent about this and also said that the skilled person would test all five). He considered structural similarity to Ψ, giving preference to those which were small incremental changes, biological origin, and the existence of methods of synthesis. Without going into all the details, this led him to reduce the list to Ψm and m1Ψ as preferred, m1acp3Ψ and m3Ψ further down because they had modifications in the Watson-Crick interface and m5D also further down by virtue of not being a pseudouridine. Dr Enright's evidence was that the information for making these decisions would come from the RNAMD. As a result, I think it is unreal to suggest that the skilled person would do the analysis proposed by Dr Enright without coming across Charette & Gray as well; the latter contains information and analysis explicitly flagged by the database and would be regarded as relatively more important.
404. I note in passing that this rejection, or at least downgrade, of m5D is hard to justify without hindsight given that the authors of UPenn had consciously included it in [0056], albeit without giving reasons.
405. Little was said by Moderna about biological origin or availability of synthetic methods but there was a good deal of evidence about the effect of, and the skilled person's thinking about, changes in the Watson-Crick interface.
406. On this topic, I think a very important point was the fact that (as both experts said in evidence and as Counsel for Pfizer/BioNTech accepted in closing) small structural changes could make a big difference in effect (an example present in both UPenn and Karikó 2008 is m6A where a small change outside the interface led to the mRNA being transcribed but not translated; there are various other similar examples in UPenn and Karikó 2008). It may be, taking Pfizer/BioNTech's case at something close to its highest, that the skilled person would see changes outside the interface as relatively less likely to cause problems, but they would still think that even a small change there could have a major or even, as Dr Enright said, "catastrophic" effect. I take on board that Prof Rosenecker was himself willing to assess the likelihood of larger or smaller changes having an effect or not, but in the main he was comparing bigger changes with smaller ones and saying that the former were more likely to have an impact. That is not inconsistent with the possibility of small changes having a big effect. He also accepted that a skilled person "could" do the experiment to see the effect of small changes outside the interface, but as with many of his answers he was just accepting the possibility, not that it was obvious.
407. I also have in mind that Dr Enright did not say that the skilled person would not make changes in the Watson-Crick interface. He said that despite the modifications potentially being catastrophic, the skilled person would still like to test them as part of the "scientific process", albeit with reduced confidence. I think this was symptomatic of his approach that negative information would be useful in gradually moving to an understanding of what was going on.
408. Pfizer/BioNTech placed some stress on methylation as a specific change. It said that methylation was known to play a role in reducing immunogenicity, from the CGK, and that if Prof Rosenecker was willing to contemplate that some small changes were unlikely to have an effect, he ought to have been willing to consider methylation at the N1 position of Ψ as a possibility. However, Prof Rosenecker was only making comparative statements based on the size of changes, and in the relevant part of his cross-examination he said that methylation was a bigger change compared to others under discussion. Further, although Pfizer/BioNTech's point about methylation might often be true, there were cases where it was not, as in m6A as tested in Karikó 2005, which was highly immunogenic.
409. Essentially absent from all of Pfizer/BioNTech's evidence, argument and analysis on these points was a consideration of expectation of success. I do not think there would be any positive expectation of success, either in general or in relation to any specific candidate change, including m1Ψ. The fundamental reason for this is that the plan of experiments proposed by Pfizer/BioNTech would be taking place in a situation where the reason for the success of Ψ was unknown and where any small change could make a big difference. The central motivation for the approach Dr Enright advocated was, at the end of the day, to try to work out why Ψ had been successful by empirical trial and error, supported by the logic that negative results would be informative, and hoping that some changes would be positive, but not having any real idea in advance which they would be. And it should not be forgotten that Dr Enright's approach had narrowed the inquiry right at the outset by going straight to the [0056] list.
410. I also bear in mind that while the effort involved in these sorts of experiments might not be huge once set up, they include both in vitro and animal model experiments, hoping that the former would limit and guide the need for the latter. So the work in its nature would not be lightly undertaken without a real, reasoned expectation of success.
411. Pfizer/BioNTech relied heavily on paragraph 40 of Prof Rosenecker's second report, which said:
40. In relation to the second step, and on the assumption that the Skilled Person had indeed turned to the RNA Modification Database, I agree that it would have been logical to search for modifications similar to Ψ and thereby arrive at the results in Dr Enright's Exhibit AJE-08, namely the entries for Ψ, Ψm, m1Ψ, m1acp3Ψ and m3Ψ. I also agree the Skilled Person would derive as much information as possible from the entries on the RNA Modification Database. However, if Dr Enright is suggesting in paragraph 6.50 that the Skilled Person would only be interested in each compound's structural similarity to Ψ, biological origin and synthesis, I disagree. If the Skilled Person was seeking to prioritise Ψm, m1Ψ, m1acp3Ψ and m3Ψ, they would also be interested in any comments or literature cited in the Database that might be relevant to the exercise.
412. He said this in the context of Karikó 2008, from which it would be necessary to go to the RNAMD to obtain the list of naturally occurring pseudouridines, but similar logic could be applied to UPenn where the list is in [0056] (plus m5D) and could be verified in the RNAMD.
413. I agree that this evidence does help Pfizer/BioNTech, but in my view only modestly, and not nearly to the extent that Pfizer/BioNTech argued. Prof Rosenecker clearly was not resiling from his view that the skilled person would not start the exercise of making modified nucleosides other than Ψ: he said as much in paragraph 39 (he also said in paragraph 38 that the skilled person would want to work out why Ψ worked before trying modifications). Nor, plainly, was he saying that there was any logic or understanding to think this shortlist would work. Paragraph 40 does not say that, and he was consistent in his oral evidence that it does not. Nor was he saying that a focus on [0056] in UPenn was justified (he was not talking about UPenn) and I do not think he meant, either, that in the context of Karikó 2008 the skilled person would be led only to this shortlist from the RNAMD. I think what he said can only assist Pfizer/BioNTech to the extent of the naturally occurring pseudouridines being a subset of modified nucleosides which potentially could be tested. He plainly did not say they would be, and this evidence said nothing about prospects of success.
414. I should mention also that Moderna argued that the skilled person would think that changing away from Ψ would risk increased toxicity, because Ψ is widely found in nature and there are metabolic pathways to deal with it. I agree this would be another factor against obviousness, but it is not central to my reasons.
415. I must assess all these matters in the round. Doing so, I find that Pfizer/BioNTech's attack of obviousness fails, and it is not a close call, either. There is no special pointer in UPenn to try other pseudouridine modifications and the focus on [0056] is artificial and hindsight-driven. But even leaving that aside, the fundamental exercise proposed by Dr Enright is one of blind trial and effort with no idea of what is likely to succeed or why, uninformed by any concrete expectation of success and without any incentive of some immediate practical application. The one concrete piece of information that the skilled person would come across in considering what to do along the lines proposed by Pfizer/BioNTech is Charette & Gray, and that is a pointer away, for reasons given above. I also prefer Moderna's position because of my finding that hindsight entered into Dr Enright's evidence.
416. None of this is to say that the sort of thing that Dr Enright proposed would be scientifically unmeritorious if a very well-resourced basic research group wanted to do it. Perhaps they would have some insight into why Ψ worked that is not present in UPenn, or perhaps they would be content to aim to publish a paper with a sort of initial SAR for Ψ. That does not make such work obvious for the notional skilled person, however. Pfizer/BioNTech submitted that if the ordinary skilled person were deterred by the uncertainty or lack of prospects of success relied on by Moderna then science would never progress. That overlooks that science may progress by people making inventions, and/or by ordinary skilled people making small incremental changes when their effects can be predicted and there is a good chance of a practical result.
417. Pfizer/BioNTech relied on the proposition that it may be obvious to do routine experiments whose results are unpredictable, citing ICOS v Actavis. At a high level, I accept the proposition, but its application is very context dependent. In ICOS v Actavis itself, the skilled person would know that there would be a minimum dose that would work and would know what experiments would be needed to identify it, but they would not be able to predict that dose and would not know it until they did the experiment. Similarly, in a situation which has come up in a number of patent cases, a skilled person might have a strong expectation that at least one of a small number of salts would work for a drug molecule, but be quite unable to predict which, so an experiment would be necessary, and obvious to do. Both those situations also involve a system which the skilled person would have a fair understanding of, although not enough to predict the final result without experiment.
418. None of this translates to the present case, where the skilled person would not understand why Ψ achieved what it did. The experiment formats proposed by Pfizer/BioNTech would be routine, but the planning as to what variants to test and according to what strategy would not be, at all. It is almost as if Pfizer/BioNTech was arguing that the fact that an experiment's results are unpredictable does not matter at all, which is plainly wrong.
419. Moderna repeatedly argued that the teaching of the prior art was so strongly in favour of Ψ that progressing it was the only way to go. I think this was overdone and presented the danger of falling into the Brugger v Medicaid trap of thinking that only the most obvious thing is obvious in law. That is incorrect. Had the upshot of the prior art been that Ψ was excellent but that there was a straightforward path to something still better (or even, just as good), then the fine results with Ψ would not have stood in the way of a finding of obviousness. Those are far from the facts, though.
420. Although Moderna overplayed this point, that does not mean there is nothing in it. It remains a relevant factor that for a skilled person with a practical interest in mRNA there was the simple and straightforward option of applying Ψ itself, with a good expectation both that it would work and that more might be learnt about Ψ. If the skilled person asked themself whether there might be other modified RNAs that would be as good or better, they would realise that a scattergun approach to finding them would be extremely uncertain and unpredictable, and that setting out to define why Ψ worked so well, so as more rationally to work on other modifications, would be a formidable research project, itself involving a lot of uncertain experiments.
421. Pfizer/BioNTech's opening submissions, both written and oral, advanced a case that the technical contribution of EP949 was only, in the sense of merely, the provision of alternative modifications to Ψ, not better ones. This was an attempt to approximate Pfizer/BioNTech's case more closely to the obviousness argument that provisionally found favour in the EPO's preliminary opinion, but did not succeed at the opposition hearing.
422. I agree with the factual basis for this argument, because as I have said when addressing the EP949 specification, it was accepted by Moderna that it does not prove superiority to Ψ (In brief submissions after this judgment was provided in draft Moderna said that it had not accepted this, but only said that it did not need to, and did not, advance a case based on superiority. Having reviewed the transcript I disagree: Moderna did accept it, albeit that the reason may have been that it felt it did not need to dispute it). However, I do not think it helps Pfizer/BioNTech, and its obviousness case still fails. The points about the skilled person, expectation of success, unpredictability and so on all apply if the skilled person were looking for alternatives to Ψ rather than improvements on it (Dr Enright's evidence was on the basis that they might be looking for either or both).
423. Moderna sought to rely on secondary evidence to support their case of non-obviousness. They relied on publications from a number of groups, all of which cited Karikó 2008.
424. In his second report, Prof Rosenecker analysed publications citing Karikó 2008 in the period between 2008 and 2015. The results fell into two categories - work undertaken by those with no apparent connection to the Karikó/Weissman group (although during their oral closing Pfizer/BioNTech suggested that three of these groups were not independent as they had input from individuals from the Karikó or Warren groups) and work undertaken by the Karikó/Weissman group itself. He pointed out that in none of them was the approach now said by Pfizer/BioNTech to be obvious, taken. In other words, none of the publications showed anyone exploring whether additional or similar benefits could be obtained from modifications other than those tested in Karikó 2008.
425. The publications covered a range of topics and Prof Rosenecker commented on them in his report in the order that they appeared in his search of the Web of Science. A particular feature, which he acknowledged, was that two of the earlier publications, Warren et al, "Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA", Cell Stem Cell 2010; 7: 618-630 ("Warren 2010") and Kormann 2011 (as discussed above) may have set the direction for the later ones.
426. In an annexe to his report he showed that all but one of what he labelled the 'independent' groups (of which there were 18 publications listed, with 16 exhibited in evidence) used and/or tested Ψ combined with m5C, which is the combination shown in Warren 2010 to give higher levels of protein expression than either used alone. All of the publications from the 'independent' groups cited either Warren 2010 or Kormann 2011, with three of the 18 citing Kormann 2011 (which itself refers to Warren 2010) and the rest citing Warren 2010.
427. So the argument may be made that the later publications do not represent independent decisions by their authors about what to do starting from Karikó 2008.
428. Moderna submitted that the secondary evidence is relevant to both UPenn and Karikó 2008. Counsel for Pfizer/BioNTech challenged whether it was relevant to UPenn as there was no evidence that any of the groups had read UPenn.
429. Pfizer/BioNTech's principal defence to this attack was that the evidence shows that Karikó 2008 was a paper that went under the radar at the relevant time. It was not widely read and its findings were not appreciated in the field when it was first published. This is an oddity in the narrative of this case. Karikó was a prominent group in the field, but as Prof Rosenecker accepted in his oral evidence, some papers do not capture the imagination of the field and it can take time for the paper to take hold in the consciousness of those in the field. The Karikó 2008 paper appears to be an example of this. Karikó 2008 was only cited in one of the publications from the 'independent' groups and that was the Warren 2010 paper, where it was cited in the context of immunogenicity. It was not cited in the Kormann 2011 paper authored by Professor Rosenecker's group, and Pfizer/BioNTech says this, along with a number of other papers published after Karikó 2008 which do not cite it, is evidence of Karikó 2008 going unnoticed by many people in the field.
430. One of the documents exhibited by Prof Rosenecker, a Nature paper from June 2015 entitled "The Billion-Dollar Biotech" comments on the Warren 2010 paper and refers to both the Karikó 2008 and Karikó 2005 papers and states that they "largely fell under the radar at the time".
431. Pfizer/BioNTech also said that an analysis of published papers can only go so far in trying to draw inferences as to what groups in the field were doing. They submit that publications do not necessarily reflect the work that was being carried out and companies active in the field may have preferred to file patents without publishing their research. Counsel for Pfizer/BioNTech pointed to Prof Rosenecker's evidence that when collaborating with CureVac, there was unwillingness to disclose details of the modified nucleotides being provided.
432. Prof Rosenecker accepted that almost all of the papers exhibited post-2013 followed the Warren 2010 or Kormann 2011 papers. The one exception is Andries et al, N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice, J Control Release 2015; Nov 10(217): 337-44 ("Andries 2015"). Andries 2015 is a paper by the De Smedt group which reports data on m1Ψ immunogenicity and translation. An earlier De Smedt group paper published in October 2010 entitled "mRNA as gene therapeutic: How to control protein expression" ("Tavernier 2010") did not cite Karikó 2005 or Karikó 2008. It was submitted by Pfizer/BioNTech that the De Smedt group may well have decided to test m1Ψ back in 2010 after seeing the Karikó 2008 work, but it took them a typical timeframe of four to five years for that work to be published.
433. I essentially agree with Pfizer/BioNTech on the secondary evidence. It is very hard to know what groups even saw Karikó 2008 and some almost certainly did not. The timing is such that it is not possible to conclude that, even if there were groups which did base their work on Karikó 2008, they would, had they worked on m1Ψ, have published by the EP949 Priority Date or soon after. There are all the usual uncertainties about other reasons why they might not have pursued the patented approach. And groups after Warren and Kormann may well have been influenced by the course they had set.
434. So I decline to give the secondary evidence any weight in Moderna's favour. This does not positively help Pfizer/BioNTech, it just means that Moderna does not get any additional benefit. Since I reject the obviousness attack on the primary evidence, it does not matter.
435. Moderna contended that there is "enormous overlap" between UPenn and Karikó 2008 and that there is no basis on which the obviousness attack starting from Karikó 2008 can succeed if the obviousness case starting from UPenn fails.
436. In closing written submissions Pfizer/BioNTech led with UPenn and kept Karikó 2008 for safety's sake, against the possibility that Moderna might critically have to rely on a point on UPenn that did not run against Karikó 2008.
437. Thus ultimately neither side made a significant positive case that Karikó 2008 was materially different. As a matter of law Karikó 2008 has to be assessed on its own merits, but since the arguments are so similar I will in this judgment aim just to explain briefly why the conclusion must be the same.
438. Karikó 2008 uses basically the same experimental approach as UPenn, but with some differences. Pfizer/BioNTech helpfully summarised it this way in closing written submissions:
240. The data in UPenn is a mixture of data from Karikó 2005 and new experimental results. At Annex A we attach a detailed comparison between the three Karikó publications.
241. Essentially, Examples 1 to 7 in UPenn are all taken from Karikó 2005 (with the exception of Figure 3E of Example 6) and Examples 8 (Fig 7A) and 11 (Fig 10B) and 12-14 overlap with Karikó 2008. Examples 9, 10 and 15 in UPenn are not found anywhere else.
242. There is unsurprisingly no overlap between Karikó 2005 and Karikó 2008. Karikó 2008 does contain material which is not found in UPenn. Karikó 2008 Figs 1, 2a, 2f, 2g, 3a, 3b, 4a and S3-S7 are not found in UPenn. Neither is any of the Discussion section.
243. We do not consider that the presence or absence of data per se would make any difference to the conclusions reached by the skilled reader as between UPenn and Karikó 2008. In both cases we submit that they would be impressed by the results with Ψ and interested to test other Ψ-like molecules.
439. The basic message of Karikó 2008 (from its Discussion section) is that "The presence of Ψ in mRNA improved its translational capacity and overall stability. It also diminished its immunogenicity in vivo." This is the same as in UPenn in the paragraphs from [00241] to [00246] quoted above.
440. In its closing skeleton, Moderna listed the differences to be the following (simplified below):
i) In Karikó 2008 there is no reference to named alternative modifications beyond those tested and the five that failed to transcribe (unlike UPenn);
ii) Karikó 2008 contains no express reference to the RNAMD;
iii) Karikó 2008 offers tentative hypotheses for the enhanced translation of Ψ-modified mRNA which Moderna contended relate to translation, not immunogenicity; and
iv) The experimental data in UPenn largely replicates Karikó 2008, but also includes all of the experimental results reported in Karikó 2005. This is correct and consistent with the fuller details quoted from Pfizer/BioNTech's closing submissions above.
441. Moderna contended that because UPenn combines Karikó 2005 and Karikó 2008, if the obviousness case starting from UPenn fails, it should not succeed over Karikó 2008 alone.
442. As to the Discussion section, Pfizer/BioNTech relied on two paragraphs bridging the left and right hand columns on page 1838 (these are the "tentative hypotheses" referred to by Moderna, a characterisation I agree with):
Our most surprising result is that mRNAs with Ψ modification have a higher translational capacity than those without modification in all tested mammalian systems. Although further studies are needed to understand the reason for this difference fully, using RIG-I(−/−) MEFs, we excluded a role for RIG-I and type I IFN in this phenomenon (Figure 3). It is possible that protein synthesis might be inhibited by RNA-dependent protein kinase activated by structural motifs present in mRNA-containing uridine17 but not Ψ modification. Consistent with this interpretation, attenuated translation of nonmodified mRNAs as compared to Ψ-modified mRNAs was observed in mammalian cells and lysates that contain RNA-dependent protein kinase but not observed in wheat germ extracts (Figures 1b and 2), which have no RNA-dependent protein kinase18.
A likely contributing factor to the enhanced translation observed with Ψ modification is an increase in biological stability of the mRNAs (Figure 4d). Indeed, higher resistance to hydrolysis by phosphodiesterases from snake venom and spleen has been reported when uridine was replaced with Ψ in dinucleotide sub- strates.19 Previous studies have also demonstrated that Ψ stabilizes RNA secondary structures by promoting base stacking,20 which could slow degradation. However, stability of mRNAs containing either uridines or pseudouridines was the same when tested by in vitro assays using human skin–associated RNases21 (data not shown). Enhanced translation might be another factor that improves stability by protecting the RNA with high ribosome occupancy.
443. In connection with them it referred to two passages from the cross-examination of Prof Rosenecker at T3/438-439 and T3/443-444. They were to similar effect and I will quote the second:
Q. Again, there is further work to be done here to investigate what it is which causes increased stability, and whether it is base stacking or not?
A. I agree.
Q. Your evidence is that the skilled person would have no reliable explanation as to why pseudouridine outperformed both uridine and the other modifications tested?
A. Correct.
Q. And you would agree that they would want to find out more about this?
A. Yes.
Q. One way of investigating this, which would occur to the skilled person, would be to try to interfere with base stacking and see if it affected stability?
A. Yes, yes.
444. In my view this is all of a piece with the arguments that I have already addressed on UPenn. The skilled person would not know what was at play and would think they needed to understand matters better before they could progress. They could initiate the sort of trial and error work put to Prof Rosenecker (the question at T3/438 was explicitly "could") to gain some enhanced understanding but their attitude to doing so would be the same as with UPenn. It may also be noted that the passage about base stacking would have to be considered by the skilled person in the light of Charette & Gray just as with the attack from UPenn, because Pfizer/BioNTech's reasoning necessarily involves going to the RNAMD.
445. I do not think these differences are material and certainly not such as to improve Pfizer/BioNTech's position; I think it was realistic to treat UPenn as its main case.
446. Karikó 2008 is in my view weaker for Pfizer/BioNTech than UPenn, for the reasons given by Moderna, in particular, as well as the lesser degree of experimental work, the omission of identifying any specific alternative modifications and of a list corresponding to [0056] (although I have not attached all that much weight to [0056] in any event). If the skilled person were however potentially interested in exploring modifications starting from Karikó 2008 then he or she might, among other things, use the RNAMD to get a list of the naturally occurring pseudouridines (and artificial ones, if any), but there is nothing in Karikó 2008 that could lead the skilled person to consider only that list as candidates (that would involve tremendous hindsight), so it would still present a diffuse and uncertain research task, for all the reasons given above.
447. There was an insufficiency pleaded as a "shepherding" squeeze. I do not think Pfizer/BioNTech ever formally abandoned it, but nor was it suggested that Moderna was taking inconsistent positions between enablement by the prior art and by the Patent, so I need say no more about it.
448. I will deal with the issues on EP565 in a conventional order: the skilled team, the CGK, the Patent itself, the prior art, added matter and novelty (which I take together because of the squeeze asserted by Pfizer/BioNTech), and then obviousness. However, I point out that significant parts of the evidence on CGK are also relevant to the skilled team, because they shed light on the actual teams in existence, and I have that in mind when I deal with the skilled team, although I cover the contents of the relevant documents in more detail when addressing CGK.
449. In addition, although I take added matter and novelty first, the issue which that leaves to the end - obviousness - is in my view the most critical and provides a clear basis to find EP565 invalid.
450. By way of sweep-up I deal with the dependent and proposed amended claims at the very end of this judgment. These have little if any significance, however.
451. There was a good deal of fencing between the parties on this issue, which tended to obscure the real issues. As is often the case, the real goal on both sides was to control the scope of the CGK by arranging the definition of the skilled team in a way which suited their respective cases. Most critically, the issue was the CGK of the skilled team in relation to the status of coronavirus vaccines: actual, in progress, and for the future.
452. I can clear away some disputes which one or other side thought the other was running but which faded away entirely (or at least very largely):
i) Although Pfizer/BioNTech thought that Moderna might be running a Schlumberger argument, that the skilled team was different for obviousness and for insufficiency, i.e. was transformed by the Patent itself, Moderna made clear that it was not.
ii) Both sides agreed that the team would include a NAV and Coronavirus Virologist, a virologist with a particular interest in coronaviruses (but this apparent agreement concealed the core dispute - see below).
iii) Both sides agreed that the skilled team would need LNP development and manufacturing expertise. Moderna pitched this at a more general level, a high level expertise which the NAV would have, and Pfizer/BioNTech elevated it to a separate team member.
453. On point (ii) above, Moderna said that while the skilled team would have a Coronavirus Virologist on board, their role would be very limited: they would be called on by the NAV only if the NAV had decided, on their own and in isolation, to work on a coronavirus vaccine. They would be asked by the NAV to identify which viral antigen should be used in such a vaccine (and it was not in dispute that they would say the S protein). Thus the Coronavirus Virologist would not, Moderna said, play any role in choosing what pathogen(s) should be the target(s) of the NAV's work.
454. Pfizer/BioNTech took the opposite extreme view. Seizing on Moderna's concession that the skilled team would have a Coronavirus Virologist as a member, Pfizer/BioNTech argued that Moderna had conceded that the skilled team would particularly know all about coronaviruses and coronavirus vaccines at the point of thinking about which pathogens to target. This was clearly not what Moderna had said, and would sell the pass on the key CGK dispute.
455. The changing scope of the dispute and the parties' preoccupation with attacking each other inhibited them from approaching the skilled team in the principled and structured way that the authorities require. Fortunately, I think the evidence enables me to do that, and the answers are quite clear.
456. First, what problem does EP565 claim to solve? The answer is an effective/improved nucleic acid vaccine for betacoronaviruses, in an LNP formulation.
457. This emerges from the claims, and the specification, including in particular the BetaCoV section from [0030], and Examples 20 to 24.
458. Second, in what established field did this problem reside at the EP565 Priority Date? On the evidence (which I address in more detail in relation to this aspect of the CGK dispute, below) while there were teams working on coronavirus vaccines, including nucleic acid vaccines, it does not seem that there were any teams working only on those pathogens. Both commercial organisations and non-governmental organisations had interests in multiple pathogens. For commercial organisations they would survey the field and then choose (largely for resource reasons) a limited number of targets to work on, and for non-governmental organisations they would look to identify which, out of many pathogens, presented the greatest need.
459. To be clear: I am not saying that non-governmental organisations were themselves the skilled addressees of EP565, but the analysis of which pathogens to target was an international collaborative effort involving industry and non-commercial organisations and it would be unreal to ignore that.
460. The problem therefore resided in the fairly broad field of vaccine development, which included both identifying and choosing pathogens to work on, and then designing, making and testing vaccines. Different commercial organisations had different vaccine platforms to deploy, but that does not bear on this facet of identifying the skilled team.
461. This all means that the skilled team would include persons with the knowledge and skill to select among the various pathogens which were the most appropriate targets. They would not know or need to know every obscure pathogen but they would know the main ones of interest, and especially viral pathogens. The virology expertise in the team would not be specifically a coronavirus expert since the established field was broader, but it would cover coronaviruses to the extent that vaccines for them were of significant interest. Since, when I come to CGK, I conclude that coronaviruses were of very significant interest, the skilled team would include someone who knew about them, among other pathogens.
462. This conclusion means that Pfizer/BioNTech is not entitled to say that the skilled team would have a dedicated coronavirus virology expert on board simply by reason of Moderna's concession; it is necessary to look at the facts. But on the facts, Pfizer/BioNTech succeeds in showing that the skilled team's expertise would extend to coronavirus virology. Moderna's position, that the NAV would not know anything material about coronaviruses, so would not choose them as a target and would never enter into a discussion with the virologist, would distort the CGK unjustifiably, as the authorities warn against. It would more or less mean that no choice of pathogen to target was ever obvious, however attractive. It is also bad on the facts, for reasons just given.
463. It is an arid debate to go into how the expertise would be distributed among imaginary members of the team, and in fact all the relevant experts before me (i.e. the NAVs and the Coronavirus Virologists, not Prof Alabi) had some appreciation of whether and why coronaviruses were thought to be important targets, and of the international discussions, so in reality the expertise probably would not be exclusively held by one or the other. Both Prof Dougan and Dr Ulmer, with broad interests in vaccinology, had SARS and MERS on their radar, in the latter case given my finding on the 2014 conference. Prof Weiss and Dr Sola were clearly not siloed away only to be consulted about which antigen to use, but had much more real world knowledge about vaccines in development and the need for them.
464. Although I have dealt with the NAV in this section, and their expertise would be focused on nucleic acid vaccines, that should not be taken to mean that they would have no knowledge of other vaccine approaches. They would have a general appreciation of other approaches so as to be able to assess whether, for a given pathogen or situation, a nucleic acid approach was the right one. I mention this in order to make clear that my conclusion on the skilled team does not per se preclude Moderna's argument that for coronaviruses, established techniques would be preferred so that it would not be obvious to apply the mRNA/LNP teaching of WO674 to coronaviruses. The argument is bad on the facts, though, for reasons given below in relation to CGK and obviousness.
465. My conclusion about the skilled team is also consistent with the specification of EP565 itself, which I deal with in more detail below, in particular in that it discusses a range of pathogens for which its "platform" could be used.
466. Finally, I think this is ultimately not a very important debate given that the main attack from the prior art is obviousness over WO674. That expressly calls out MERS-CoV and "another key vaccine" for it, in Example 20. Of course, WO674 talks about other pathogens too, notably flu, but even if there were a skilled team who knew very little about coronaviruses they would inevitably look into them to some extent in deciding what, if anything, to progress from WO674 and I am sure that would readily lead to the sort of materials that Pfizer/BioNTech argues, and I accept, were CGK about the need for coronavirus vaccines.
467. I will set out the agreed CGK and then deal with the two remaining areas of dispute. The agreed CGK is based on the document prepared by the parties. I have edited it down, but that does not mean that what I have removed is not CGK.
468. The agreed document refers to the Priority Date. It was prepared by the parties before Moderna gave up on priority entitlement. I do not consider there was any material change in the CGK in the period between the EP565 Priority Date and the EP565 Filing Date in 2016 (as, for example, Dr Sola said; I have rejected above one small aspect in which at one point in her evidence she suggested otherwise).
Coronaviruses - Overview
Classification of coronaviruses
469. Coronaviruses are viruses included in the large subfamily Orthocoronavirinae, within the Coronaviridae family and Nidovirales order.
470. Coronaviruses can infect a range of animal species, including humans, other mammals and birds. The majority of coronaviruses cause mild to severe enteric or respiratory disease, but some also cause systemic disease and can affect the central nervous system.
471. Human coronaviruses were first identified in the 1960s. By the 1980s, there was an established field of scientific research relating to the study of coronavirus pathogenesis. The focus of this research related to understanding the molecular biology of coronavirus replication, as well as disease mechanisms and prevention in animals, until the SARS outbreak of 2002 - 2003, which resulted in an increase of interest in the study of human coronaviruses.
472. Coronaviruses can be subdivided into four genera: (i) alphacoronaviruses; (ii) betacoronaviruses; (iii) gammacoronaviruses; and (iv) deltacoronaviruses. In general, human coronaviruses are found in the alpha and betacoronavirus genera. At the EP565 Priority Date, there were four betacoronaviruses known to infect humans: (i) human coronavirus OC43 (HCoV-OC43); (ii) human coronavirus HKU1 (HCoV-HKU1); (iii) SARS-CoV; and (iv) MERS-CoV.
473. Alphacoronaviruses and betacoronaviruses are typically thought to originate in bats with intermediary species as they evolve to zoonotic infection (i.e., have the capability to spread to humans). Dromedary camels are persistent reservoirs for MERS-CoV and the source of zoonotic infection.
Coronavirus structure and genome
474. Coronavirus virions are enveloped, roughly spherical viruses, with diameters of between approximately 80 to 125 nm. All coronaviruses have the same morphological configuration. They have positive-sense, single-stranded, polyadenylated genome RNAs complexed with nucleocapsid (N) protein, surrounded by a lipid envelope. Positive sense means that their genetic material is stored as RNA that can be immediately translated by ribosomes inside a host cell.
475. In addition to the N protein, which covers and packages the entire RNA genome to build the viral ribonucleoprotein, or RNA-protein complex, at the core of the virus, the other key structural proteins of coronaviruses include:
a) the spike (S) protein (see further below);
b) the membrane (M) protein - this is smaller than the S protein and spans the viral membrane. It is essential for viral assembly and interacts with the S, E and N proteins and RNA to form the viral particle; and
c) the envelope or small membrane (E) protein - this is a small protein that sits in the viral membrane. It is abundantly expressed in infected cells and plays a role in viral assembly and particle formation,
which are embedded within or span the lipid membrane.
476. The following steps occur in the lifecycle of a coronavirus:
a) cell entry - different coronaviruses target different cell surface receptors, typically within organs of the respiratory and intestinal systems, to allow entry from the cellular environment. Coronaviruses bind to their specific cell surface receptor via the S protein, which triggers fusion of the viral lipid membrane with the cell membranes and allows the viral nucleocapsid (RNA and protein coat) to enter the cell and initiate viral replication. The S1 subunit, at the tip of the protruding spike, binds the receptor before detaching from the S2 subunit, which then folds to pull the two membranes together and cause fusion;
b) translation - once inside the cell, ribosomes will begin to translate the viral replicase gene of the genomic RNA into the proteins required for viral RNA synthesis;
c) RNA replication - replication of the whole viral genome takes place. The 'positive' RNA strand is copied into a 'negative' complementary strand, which is itself used as a template to produce more positive strands;
d) transcription - sub-genomic mRNAs encoding the non-replicase viral proteins are transcribed before being translated; and
e) assembly or formation of viral particles - the translated structural proteins accumulate, and the RNA genome complexes with N protein. This ribonucleoprotein comes together with E and M proteins, which are embedded in intracellular membranes, to package the genome into the viral particle. S protein incorporates in the membrane to form complete viral particles that are then transported out of and released from the cell.
477. Coronaviruses have also evolved methods to interfere with the cellular signalling pathways associated with the innate immune response that are triggered when the cell recognises viral RNA. For example, they produce proteins that block signalling and the activation of signalling genes.
Spike Protein (S)
478. The S protein is the largest glycoprotein (the S protein is a glycoprotein because it has oligosaccharide chains covalently attached to its amino acid sidechains) on the virion surface and mediates cell receptor binding and membrane fusion during cell entry. It is trimeric, i.e., formed of three identical copies of the same S protein associated together. Each S protein consists of: 1) an S1 subunit, which forms the globular head of the S protein and attaches to host cell surface receptors; and 2) an S2 subunit, which forms the transmembrane stalk of the S protein and is responsible for fusion between the virion and the target cell plasma or endosomal membrane.
479. The sequence of S proteins is variable across genera and species of coronaviruses as the determinant of host specificity. The S2 subunit is the most conserved region of the S protein, meaning that there is less genetic variation between the S2 subunits of different coronaviruses than occurs in the S1 subunit. In contrast, there is often more divergence in the genetic sequence of the S1 subunit as between coronavirus species. The S1 subunit includes a region called the receptor binding domain (RBD), which is the most variable part of the coronavirus genome.
480. The role of the S protein in attachment and fusion means that it plays a crucial role in determining: (i) the animal species which a coronavirus can infect; (ii) the cell type within that host which can be used for viral replication; and (iii) the virulence of the virus.
481. The S protein is known to be the main inducer of neutralising antibodies in the host following coronavirus infection for all known coronaviruses.
Diseases caused by human betacoronaviruses
482. The betacoronaviruses HCoV-OC43 and HCoV-HKU1 are known to cause human respiratory disease, but the resultant disease is generally mild except in immunocompromised individuals.
483. By the EP565 Priority Date, there had been two serious human betacoronavirus outbreaks: SARS-CoV (2002 - 2003) and MERS-CoV (from 2012). SARS-CoV causes SARS (Severe Acute Respiratory Syndrome), a contagious disease typically presenting a triphasic pattern of symptoms progressing from an initial fever and cough to shortness of breath (dyspnea) and hypoxia, frequently accompanied by diarrhoea and often requiring mechanical ventilation. MERS-CoV causes MERS (Middle Eastern Respiratory Syndrome) and typically affects the lower respiratory tract of humans, with potential to cause severe pneumonia, often accompanied by renal failure.
The 2002-2003 SARS outbreaks
484. SARS-CoV was identified in China in 2003 and was determined to have spread since 2002, causing severe respiratory disease and death in humans. By late March 2003, the novel coronavirus SARS-CoV had been identified in SARS patients and the mapping of its genome was completed in April 2003.
485. The SARS-CoV outbreak was largely contained by the end of 2003.
MERS outbreaks
486. MERS-CoV cases were first reported in the Middle East in 2012. Transmission was mainly to humans from infected dromedary camels (which are a 'reservoir' for the virus). MERS-CoV has a higher mortality rate than SARS-CoV but, unlike SARS-CoV, does not spread easily between humans.
487. The majority of MERS-CoV cases occurred in the Middle East, with sporadic cases reported in various European countries and an outbreak in South Korea in 2015.
SARS-CoV and MERS-CoV neutralising antibodies and animal models
488. To evaluate the production of neutralising antibodies against a betacoronavirus, the Coronavirus Virologist would expect the following studies to be performed:
a) first, it must be established that antibodies that bind specifically to the viral antigen(s) are generated. An ELISA assay can be used to measure the antibody titre generated against one or more betacoronavirus antigens; and
b) next, it must be established whether the antibodies generated are neutralising, i.e., whether they have the capacity to prevent the virus from infecting additional cells. Neutralisation assays are performed in cell culture using sera from immunised animals.
489. In general, an animal model should be representative, i.e., reproduce the disease occurring in the natural host reliably and the animal should be susceptible to the coronavirus in question, ideally exhibiting lethal infection. Wild type mice are not representative of MERS or SARS, so specific animal models had to be developed.
490. For SARS-CoV, animal models included:
a) mice with adapted virus - researchers used a passaged SARS-CoV strain to study vaccine candidates in mice, which are not naturally susceptible to severe disease. The virus had been passaged, i.e., grown and replicated over many generations, in the lungs of mice, until a mutant strain had developed that was adapted to the mice and caused severe disease signs and lung pathology;
b) transgenic mice - mice were genetically modified to overexpress the SARS-CoV receptor, which produced a lethal model, but was overly sensitive as receptors were expressed even in the brain; and
c) ferrets and non-human primates - ferrets and non-human primates were also used to study SARS-CoV, though it was difficult to secure funding to use non-human primates by the EP565 Priority Date.
491. There were limited animal models available to study MERS-CoV and those that did exist were imperfect. The available options included:
a) rabbits - rabbits are susceptible to MERS-CoV, however they do not exhibit signs of severe disease and this is not a lethal model. MERS-CoV can replicate in the respiratory tract, which can be measured;
b) transgenic mice - Mice are not naturally susceptible to MERS-CoV. Genetically modified mice which were susceptible to infection had been developed as a lethal model, first described in April 2015. However, this model was not commercially available by the EP565 Priority Date;
c) sensitised mice - these were created by administering adenoviruses rendering the mice temporarily susceptible to infection. This model had some limitations but was used before the transgenic mice model became available and was first published in 2014;
d) marmosets - showed severe lung disease, similar to humans. However, as primates, marmosets require special licensing and facilities and were not commonly used; and
e) camels - as the original host animal for MERS-CoV, camels are infected, but do not show severe disease. They had been used to evaluate a vaccine designed to prevent transmission to humans. However, very few labs had access to camels as they are impractical to house for research purposes.
The Immune System
492. The immune system is a complex network of organs, cells and proteins that protects the body from harmful substances, pathological cellular changes and disease-causing agents (such as bacteria, viruses, fungi and parasites).
493. In mammals, the immune system has two key subsystems:
a) the innate immune system, which is an immediate and non-specific system that recognises and reacts to certain patterns and foreign molecules found in broad categories of pathogens; and
b) the adaptive immune system, which is initially activated by the innate immune system, and provides a highly tailored antigen-specific response against epitopes found in a particular pathogen. When the adaptive immune system is exposed to a new pathogen, there is a lag time between the initial exposure and the maximal response, during which time the adaptive immune response matures through an iterative process. Upon subsequent exposure to the pathogen, an antigen-specific memory immune response is recalled enabling it to recognise and respond to the relevant antigens quickly.
494. The adaptive immune response is mediated predominantly by:
a) B cells and antibodies or immunoglobulins (Ig) - antibodies are proteins produced by circulating B lymphocyte cells which bind a specific antigen (i) on the pathogen, thereby interfering with its ability to enter and infect cells (neutralisation); or (ii) on the exterior of infected cells to mark them for destruction (this is also known as the humoral response). Further effects of this binding may be (i) activation of the complement system; or (ii) phagocytosis of the detected pathogen; and
b) T cells – once T lymphocyte cells recognise epitopes within a specific antigen that are presented on the surface of infected cells and either (i) kill infected cells (by CD8+, or cytotoxic or killer T cells); or (ii) support the B cell and antibody response through production of cellular activation signals called cytokines or chemokines (by CD4+, or helper T cells) (this is also known as the cellular response).
Vaccines - Overview
495. A vaccine for an infectious disease is a biological preparation which induces an active immune response which is specific to a pathogen. Such a preparation or biological composition is designed to stimulate an immune response against a specific pathogen, triggering the formation of immunological memory against it, such that the vaccinated individual can mount an effective immune response upon subsequent exposure to the pathogen, thus protecting the individual from disease. In certain cases, a vaccine may also be intended to help the individual to clear existing disease.
496. To achieve this, the recipient of the vaccine must be exposed to a pathogen (or antigens from it) in such a manner that an immunising effect is achieved. Vaccines typically contain antigens, which are molecules or substances that can be recognised as foreign or non-self by the relevant components of the immune system, and which are typically derived from the target pathogen (or, alternatively, a whole pathogen particle). Components of the immune system may be able to interact with multiple sites on an antigen, known as epitopes.
497. Any antigen that triggers an antigen-specific immune response is known as an immunogen. All immunogens are antigens but not all antigens will be immunogens, as not all antigens may be able to induce an immune response. Vaccines must be immunogenic and generate a specific response against the antigen(s) they contain.
Vaccination
498. Upon vaccination (or natural infection), the innate immune response is initially activated and stimulates the subsequent adaptive response against the specific antigen(s) in the vaccine and begins the process of developing immunity. Immunity conferred in this way should culminate in the production of memory cells and, therefore, confer immunity on the patient. This is described as active immunisation.
499. By contrast, passive immunity occurs with the transfer of pre-existing antibodies to an unimmunised individual. Unlike active immunisation, passive immunisation does not result in any immunological memory and the immunity conferred will last only as long as the transferred antibodies survive (although the individual may also be developing their own active immune response in parallel).
500. The theoretical goal of vaccine development is sterilising immunity, which is complete protection against initial infection. However, this is uncommonly achieved, as it requires high levels of highly specific antibodies against the pathogen to eliminate the pathogen before it can replicate, ideally at the entry site.
501. More commonly, vaccines achieve a level of immunity which protects against severe disease, rather than infection per se. This may be mediated by antibodies that can limit the number of cells infected by the pathogen and reduce the burden of disease. In other cases, CD4 and CD8 T cells limit the magnitude and duration of disease by facilitating the destruction of infected cells and clearing the infection faster.
Types of vaccine
502. The following types of vaccines were commercially available at the EP565 Priority Date:
a) Inactivated vaccines – whole-pathogen vaccines in which an infectious agent has been 'killed' or inactivated, for example, via heat, chemicals or irradiation, before use in a vaccine. Inactivated pathogens cannot replicate, and so repeated doses of inactivated vaccines are often required for immunity to be achieved.
b) Live attenuated virus vaccines - whole-pathogen vaccines that are commonly derived from an infectious agent that has been repeatedly passaged in non-human cells until it has become less virulent in humans or genetically modified to decrease virulence (that is, less likely to cause disease). As these vaccines use actual live viruses, the response they induce mirrors that of an infection - i.e., triggering of the innate immune system, antigen presentation and a T and B cell response followed by the formation of an immunological T and B cell memory.
c) Virus-like particle (VLP) vaccines - contain one or more antigens which interact and / or self-organise into particles resembling a virus of interest (but typically do not contain genetic material required for viral replication).
d) Subunit vaccines - contain an antigenic component of the infectious agent and (usually) an adjuvant to stimulate the immune response. These antigen subunits can be based on protein or polysaccharides. Some subunit vaccines were produced using recombinant technology, which has been in use for vaccine production since the 1980s.
503. In addition, the following types of vaccine were being investigated and had progressed to human trials by the EP565 Priority Date:
a) Viral vector vaccines - use modified viruses (either replicating or non-replicating) to deliver genetic information from an infectious agent of interest into cells. Viral vector platforms use modified viruses whose own genome has been altered such that the virus cannot cause disease in the host. The role of the modified virus is to infect the target cells and instruct them to make the antigen according to the sequence of the nucleic acid which is being delivered. Recombinant viral vectors (for example, adenovirus or modified vaccinia Ankara virus) had been investigated.
b) DNA vaccines - DNA encoding an antigen (or antigens) from an infectious agent of interest is delivered to cells, transcribed, and translated to produce antigens against which an immune response is generated.
c) RNA vaccines – a small number of clinical trials for mRNA vaccines were ongoing in 2015. However, the details, data and any outcomes from those trials would not have been CGK in 2015.
504. Nucleic acid vaccines contain antigen genes encoded by either DNA or RNA. They make use of the cellular processes described above to produce the antigen in cells of the vaccinated individual / animal with the aim of generating an antigen-specific immune response in a manner similar to what happens during a viral infection.
Vaccine Design
505. There are several technical aspects to consider when designing a vaccine, including the following:
a) Antigen design - the antigen(s) chosen will typically be part(s) of the infectious agent of interest and should preferably stimulate the type of immune response which is induced by natural infection. Important considerations include the choice of antigen from the many present in the pathogen and the 3-dimensional conformation of the antigen (in particular for induction of antibody responses).
b) Adjuvant - for some vaccine types an adjuvant may be required to help stimulate the immune system to respond to an antigen which produces an inadequate response on its own. An adjuvant is a substance added to a vaccine to improve, or modulate, the type of immune response that is generated. While adjuvants can be classified in various ways, in general they work by activating the innate immune response, for example through the stimulation of pattern recognition receptors. Various adjuvant substances had been developed and approved for human use by the EP565 Priority Date. For example, alum (aluminium compounds) and MF59 were being used / investigated for co-administration with an active ingredient. Certain lipids such as cationic lipids were also known to have an adjuvant effect.
c) Delivery - certain vaccine types, such as viral vector and nucleic acid vaccines, use the cellular machinery to produce antigens, and therefore require delivery into cells in order to be effective. For these vaccines, the formulation of the vaccine must facilitate delivery to the relevant cells.
d) Administration route - possible administration routes for vaccines include intramuscular, intravenous, intradermal and subcutaneous injection (into the muscle, veins, skin and under the skin, respectively), as well as oral, intraocular, intratracheal and intranasal (via the mouth, eye, trachea and nose, respectively) vaccination and inhalation. In general, the administration routes chosen are low risk.
e) Dosing – the dose of vaccine administered, as well as the dosing scheme (i.e., intervals and frequency) and age at which recipients are vaccinated must be considered.
f) Toxicity / side effects - a vaccine must be stable and effective but must not cause too high a level of activation of the immune system leading to toxicity or side effects (either local or systemic), particularly if it is to be administered to a healthy population. In addition, immunopathology or the potential for enhancement of disease should be avoided.
g) Cost – in order for a vaccine to meet the needs of the target patient population, it must be capable of being scaled up for production at an appropriate cost.
Evaluating vaccines
506. Evaluating the effectiveness of a vaccine requires in vivo testing. For some pathogens, for example influenza, there are established 'correlates of protection', which indicate an immune response that is responsible for and statistically correlates with protection at defined amounts as measured by a standardised assay. Where a quantitative correlate has been established, this may permit the prediction of protection from animal studies or a small Phase I human clinical trial - for example, in the case of influenza virus a serum haemagglutination inhibition antibody titer in humans of about 1:40 is considered protective. Where no correlate of protection exists, demonstration of protection can be established by challenge experiments, including in a suitable animal model where animals are exposed to live pathogen to assess whether a vaccine can confer protection against infection and / or disease caused by the pathogen.
507. In the course of developing a candidate vaccine, a large number of tests will be performed, and the laboratory or group developing the vaccine will have established in advance the criteria for success / failure at each stage. Depending on the type of vaccine, those steps would likely include:
a) in vitro studies to characterise the purity and quality of the vaccine components, such as various analytical methods to separate the individual components and assays to determine their identity;
b) in the case of nucleic acid vaccines, testing for effective delivery to and expression in cells;
c) assessing the level of immune response elicited in an appropriate animal model, and that such response is specific to the antigen of interest;
d) establishing that the immune response is neutralising, in the case of an antibody response;
e) testing the candidate vaccine in a suitable animal challenge experiment to establish whether it confers protection to the target disease;
f) assessing tolerability and safety of the candidate vaccine;
g) testing the stability of the candidate vaccine (and the formulation components, where relevant);
h) developing methods to consistently produce and characterise the vaccine and its components; and
i) considering the complexities of large-scale manufacture relating to that specific candidate.
508. At the EP565 Priority Date, various methods were used to assess antibody and T cell responses to vaccination. Assays routinely used to measure the antibody immune response included:
a) ELISA assay - used to measure serum antibody levels. Broadly speaking, a higher antibody titre - particularly an IgG titre, which is more antigen-specific - indicates a more robust adaptive immune response.
b) Neutralisation assays - used to measure serum neutralising antibody levels. In the context of viruses, neutralising antibodies are those which block viral infectivity by stopping the ability of the virus to: (i) bind to the cell; (ii) replicate within the cell; and / or (iii) exit from the cell.
509. Challenge and safety studies generally involve vaccinated animals being exposed to doses of the infectious agent that cause measurable levels of infection, disease or death and the magnitude of these parameters is assessed.
510. Ultimately, a vaccine must also be tested in the natural host to determine its safety and efficacy. What is required will depend on all the circumstances but there will need to be sufficient basis for concluding that a vaccine candidate can be safely administered to otherwise healthy individuals before clinical trials can take place.
Nucleic Acids
511. Although the priority dates and relative focuses of EP949 and EP565 are different, at the level of detail relevant there is no difference in the agreed CGK on the basics of nucleic acids, so I refer back to the section on them in the EP949 agreed CGK at paragraphs 162 to 182.
Nucleic acid vaccines
512. Nucleic acid vaccines contain DNA or RNA encoding the antigen(s) of interest. They make use of cellular processes to produce the antigen in cells of the vaccinated recipient (animal / individual) with the aim of generating an antigen-specific immune response in a manner similar to that which occurs during a viral infection. Rather than exposing the recipient to an antigen directly upon injection, the nucleic acid encoding an antigen from the infectious agent of interest is administered to the recipient and delivered to a cell. Where DNA is used, it has to reach the nucleus of a cell so that the encoded antigen sequence can be transcribed into mRNA. That mRNA is then transported back out of the nucleus into the cell cytoplasm where translation takes place. RNA vaccines, on the other hand, need only be delivered into the target cell cytoplasm for translation and production of the encoded antigen. The cell then presents the antigen to the immune system, which stimulates an antigen-specific response that should ultimately protect the recipient upon future exposure to a pathogen containing the antigen.
513. Throughout the 2000s and before the EP565 Priority Date there was interest in the development of nucleic acid-based vaccines. By the EP565 Priority Date, it was well known that nucleic acid vaccines had the potential to be:
a) safe - they do not involve living organisms and do not have the risk of reversion to a virulent state. They also are highly purified and may not require the use of adjuvants.
b) effective - they express antigens in situ over a period of days to weeks priming potent immune responses and have the potential to generate both humoral and cellular immune responses, such as cytotoxic T-lymphocyte responses. In the case of antibody responses, the antigen retains a native conformation similar to that produced by infection with the pathogen. In the case of T cell responses, production of antigens in situ is a more efficient means to induce T cells.
c) focused - the immune response can be directed towards a selected antigen of interest, rather than all antigens in the organism.
d) adaptable - the technology uses conventional materials and equipment and manufacturing processes that can quickly be adapted to a new antigen target, and therefore is amenable to rapid response.
514. Nucleic acid vaccines also have the potential to generate both humoral and cellular immune responses, such as cytotoxic T-lymphocyte responses. However, despite these potential advantages, by the EP565 Priority Date no nucleic acid vaccine had been approved for human use.
DNA Vaccines
515. Early work in the 1990s suggested that both DNA and RNA were capable of expressing encoded genes in animals and could therefore potentially be used as nucleic acid vaccines. However, initially the field focused on DNA. This was primarily due to concerns about the instability of RNA, and the high cost of manufacturing RNA vaccines, as exogenous RNA is susceptible to degradation by enzymes known as RNases. It was therefore initially assumed that feasibility of manufacturing would be challenging and thought that high doses of RNA would be needed as compared to DNA. In contrast, DNA plasmids were relatively easy and inexpensive to produce, albeit, not using GMP procedures (GMP stands for Good Manufacturing Practice and is an essential system for ensuring that products are consistently produced and controlled according to quality standards).
516. Initial success with DNA in small animal models led to high expectations around the potential of DNA-based vaccines. However, issues concerning the use of DNA for vaccination had become apparent, including that the efficacy of DNA vaccines did not scale from small animal in vivo models to humans, demonstrating poor immunogenicity in early clinical trials. Very high doses of DNA had been shown to be required for an immune response in humans, which would be impractical as a matter of production, and increase the risk of toxicity and / or side effects.
517. The disappointing results in humans led to research activity directed towards the optimisation of DNA constructs, the use of co-expressed adjuvants to target the innate immune system and increase immunogenicity (thus reducing the required dose), and improved delivery methods to overcome the barriers to efficient transfection. However, other groups moved their focus from DNA to alternative approaches (including RNA). By the EP565 Priority Date, a number of teams were working on nucleic acid vaccines, with the majority focusing on RNA vaccines.
518. At the EP565 Priority Date, there were a number of known differences between DNA and RNA in the context of vaccines, including the following:
a) Delivery - both DNA and RNA must cross the cell membrane into the cytoplasm to function. However, DNA must additionally cross the nuclear membrane and enter the nucleus before it can be transcribed. The transcribed mRNA must then exit the nucleus to be translated in the cytoplasm. This poses an additional delivery hurdle that must be overcome for DNA. RNA vaccines, on the other hand, need only be delivered into the target cell cytoplasm for translation of the encoded antigenic sequence.
b) Stability - DNA is typically more stable and less susceptible to nucleases than RNA, owing to its double-stranded structure and deoxyribose sugar.
c) Antigen expression kinetics - antigen expression and decay appear more rapid for RNA, as compared to DNA where antigen expression can persist for weeks.
d) Safety - there is no risk that RNA can integrate into the genome of the immunised host and disrupt the existing genetic material, as it does not enter the nucleus and does not integrate into the DNA genome.
e) Size - DNA is typically larger than the equivalent gene delivered as RNA.
f) Host cell interactions - both DNA and RNA contain patterns recognised by the innate immune system, but DNA is less immunostimulatory than RNA. Certain RNA molecules can trigger innate immune responses. This potentially results in enhanced immunogenicity which is potentially beneficial for use in vaccines.
RNA vaccines
519. At the EP565 Priority Date, the two main forms of RNA vaccine being investigated were: (i) vaccines using self-amplifying mRNA molecules; and (ii) vaccines using non-self-amplifying/conventional mRNA molecules. There were advantages and limitations of each approach, and most RNA vaccine research groups were focused on one or the other. As I indicate below, there was at one point an issue about the meaning and scope of the claims of EP565 as relates to the difference between these two forms of RNA vaccine, and the existence of that dispute led the parties to frame this part of the agreed CGK text with two alternative forms of wording. The claim interpretation issue fell away and I have resolved the form of expression of this section by referring to "conventional mRNA" and "non-self-amplifying" mRNA vaccines. This makes no difference to the substance and does not mean that I have purported to resolve the claim interpretation issue. It is just for convenience.
Non-self-amplifying/conventional mRNA optimisation
520. RNA molecules used within non-self-amplifying/conventional mRNA vaccines (and other therapeutics) primarily consist of five main components:
a) a 5' cap: this facilitates recognition of mRNA and can improve translation initiation. It protects mRNA from degradation and increases RNA stability during protein synthesis;
b) a 5' untranslated region (5'UTR): this is a non-coding region that precedes the ORF encoding the protein of interest. It plays a role in translation efficiency and can affect the stability of the mRNA;
c) an ORF: this encodes a protein (such as an antigen);
d) a 3' untranslated region (3'UTR): this is a non-coding region that follows the protein of interest. It plays a role in regulation of mRNA-based processes including stability and translation of the mRNA; and
e) a poly(A) tail at the 3' end which increases stability of the mRNA. In general, the longer the tail the more stable the molecule, up to a point (around 60-120 nucleotides). It also aids in transcription termination, export of the mRNA from the nucleus, and translation.
521. These elements can be optimised to improve the stability and translation of the non-self-amplifying/conventional mRNA molecule by some or all of the following techniques:
a) the cap structure can be modified to improve stability and the efficiency of protein translation;
b) the choice of 5'UTR and 3'UTR sequence can affect the stability of the molecule and increase protein translation;
c) codon optimisation can increase translation efficiency;
d) length of the poly A tail can play a role in protein expression; and
e) purification may decrease immune activation and can potentially increase translation.
522. In addition to the modification of the elements outlined above, by the EP565 Priority Date, it had been shown that the incorporation of modified bases, such as pseudouridine, into non-self-amplifying / conventional mRNA could lead to increased expression of therapeutic proteins in vivo.
523. With respect to the immunogenicity of self-amplifying [m]RNA and non-self-amplifying / conventional mRNA in the vaccine context, it was understood that while a degree of direct innate immune activation by an RNA vaccine construct was potentially desirable to assist with the generation of an adaptive immune response, if a significant innate immune response were triggered, it could interfere with translation.
Self-amplifying mRNA vs non-self-amplifying/conventional mRNA vaccines
524. Self-amplifying mRNA encodes, in addition to the gene of interest, protein(s) that enable its replication. To achieve this, an engineered RNA virus-derived replicon is used, the most common of which are derived from alphaviruses. An alphavirus-based replicon encodes four non-structural proteins, a sub-genomic promoter and the protein of interest (which replaces the coding sequences for the alphavirus structural proteins). Encoding the additional replicase protein(s) means that self-amplifying mRNA is approximately five times larger than non-self-amplifying/conventional mRNA encoding the same protein of interest.
525. The four non-structural proteins are translated upon entry into the cell, and form a complex called RNA-dependent RNA polymerase. This complex then enzymatically creates copies of the replicon via a double-stranded intermediate in an amplification process. This leads to high copy numbers of mRNA encoding the gene of interest.
526. At least the following points from the review article Deering 2014 would have been CGK by the EP565 Priority Date:
a) initial concerns regarding the stability and large-scale manufacturing of RNA therapeutics were no longer perceived as barriers to implementation. Manufacturing of RNA had reached the scale and quality sufficient for Phase I human clinical trials;
b) a variety of different delivery approaches had been utilised to deliver both self-amplifying mRNA and non-self-amplifying/conventional mRNA vaccines;
c) there was a balance to be struck between expression of the antigen encoded by the RNA and activation of the innate immune system; and
d) there were two main forms of RNA vaccine being investigated - self-amplifying mRNA and non-self-amplifying/conventional mRNA.
RNA vaccine targets
527. While the early work on RNA vaccines focused on cancer targets due to the considerable unmet medical need, by the EP565 Priority Date infectious disease targets were also under investigation.
Vaccine associated enhancement of disease
528. It was known that in some instances, vaccines could serve to enhance or potentiate disease, rather than prevent it, on subsequent infection. Vaccine-associated enhancement of disease (VAED) is an umbrella term that includes antibody-dependent enhancement (ADE), antibody-enhanced disease (AED) and Th2-mediated (eosinophilic) pathology. At a very high-level: (i) ADE occurs when non-neutralising antibodies increase the ability of a virus to infect cells; (ii) AED involves the over-production of inflammatory cytokines and chemokines resulting in a pathological level of inflammation; and (iii) a pathogenic Th2 response results in eosinophilic tissue infiltration. VAED is a phenomenon that can occur for multiple vaccine targets, and in relation to various vaccine technologies, and is relevant to vaccine development generally. A particularly well-known example of this was the experience of enhanced disease in children vaccinated with formalin-inactivated RSV vaccine in clinical trials in the 1960s. ADE was also an issue during the development of dengue vaccines.
Coronavirus Vaccine Development at the EP565 Priority Date
529. The SARS-CoV and MERS-CoV outbreaks had demonstrated the zoonotic potential of coronaviruses and their ability to cause severe disease and death in humans.
530. By the EP565 Priority Date, a number of SARS-CoV and MERS-CoV vaccine candidates had been investigated but no vaccine had been authorised for either SARS-CoV or MERS-CoV.
531. While it was not the only approach, the antigens used in most candidate vaccines for MERS and SARS were based on the S protein of those viruses. The S protein was known to be the main inducer of neutralising antibodies following infection. This is the case for all known coronaviruses.
Delivery of Nucleic Acids
532. The in vivo delivery of naked exogenous nucleic acids such as DNA and RNA means that such nucleic acids are not associated with a carrier and / or are formulated in buffer only.
533. Cellular entry is a prerequisite for any desired therapeutic effect.
534. Where the nucleic acid molecule being delivered is DNA, it must also cross the nuclear membrane to be transcribed into RNA. That RNA must then be transported back to the cytosol to be translated into protein. Nucleic acids do not easily cross nuclear membranes unless the cells are dividing. Transfection is the process of artificially introducing nucleic acids into eukaryotic cells, using various chemical or physical methods. An effective nucleic acid delivery system must carry the nucleic acid into the targeted cells, to avoid degradation and ensure its transcription and / or translation.
535. Another key issue for the delivery of naked exogenous nucleic acids is that they are not very stable in vivo and have a relatively short half-life. In particular, endonucleases in the body degrade nucleic acid chains into smaller components. A significant proportion of nucleic acids delivered without a suitable carrier system will likely be degraded before they can reach the target cell(s). Different classes of nucleases exist - for example, deoxyribonucleases (DNases) will break down DNA and ribonucleases (RNases) will break down RNA.
536. Early administration of DNA and RNA vaccines in mice used naked forms of DNA / RNA. Although this approach was shown to lead to an immune response, degradation likely limited the amount of DNA / RNA internalised by the cells, and the resulting amount of antigen expressed.
537. While individual nucleic acid delivery systems differ, most have at least the following features in common:
a) a complex is formed between the nucleic acid and components of the formulation. The complex can, in many cases, protect the nucleic acid from exposure to degradation by nucleases, thereby increasing the nucleic acid's stability and half-life. This can indirectly facilitate uptake by the cell, as the nucleic acid is not degraded before getting into the cell. If the delivery system contains cationic components, these will interact electrostatically with the negatively charged nucleic acids to form the complex;
b) formulations also form particulate structures. These are typically roughly spherical which, by their nature, facilitate uptake by cells via one or more endocytic pathways; and
c) particles are internalised by cells via small membrane-bound compartments (vesicles, endosomes or phagosomes formed from membrane invagination), but the nucleic acid must be released into the cytoplasm to take effect. For formulations with a cationic element, it was thought that this element may facilitate destabilisation of the compartment membrane, thereby releasing the nucleic acid into the cytoplasm.
538. The nature of the interaction and type of complex formed between the nucleic acid and formulation components will differ according to the delivery system.
Design of delivery systems for nucleic acids
539. There are several factors to consider when designing, selecting or optimising a formulation for use in a nucleic acid vaccine. These factors would vary according to both the nucleic acid and the purpose but would include the following:
a) compatibility of the components or conditions of the process generating the formulation, with the nucleic acid. The overriding principle is that the integrity and functionality of the nucleic acid must be preserved;
b) efficiency of functional nucleic acid delivery into cells in vivo, including in animal models. The size of the nucleic acid may affect the amount of delivery at a molar level, i.e., many smaller molecules may be delivered for one larger molecule;
c) ability of the formulation to dissociate from the nucleic acid after intracellular delivery;
d) stability and degradation of the nucleic acid and formulation components, both separately and together in the co-formulated drug product;
e) biodistribution of the vaccine. While the precise location of gene expression is not as important for a vaccine as compared to, for example, gene therapy requiring delivery to a specific cell type, the formulation may affect how much, when and where material is produced;
f) potential off-target effects that could have an impact on the functionality of the nucleic acid, such as interference with translation resulting in lower levels of antigen production and lower vaccine potency; and
g) logistical considerations. For example, the reproducibility, cost and efficiency of the manufacturing process, the availability of raw materials, the ability to scale up the process and any safety concerns.
Lipid-based carriers for nucleic acid delivery
540. By the EP565 Priority Date, a variety of lipid (or lipidoid) carriers had been developed to deliver a range of nucleic acids, including DNA and RNA, for a variety of purposes, including gene therapy, protein replacement and vaccine applications. Naturally occurring lipids usually have a single hydrophilic head group and one or two hydrophobic tail groups. Lipidoids are synthetic lipid-like materials which are capable of rapid, parallel generation and have similar properties to lipids, but may have more than two lipid tails as they can be formed with large multifunctional head groups. Whole libraries of lipidoids had been developed by the EP565 Priority Date and they were frequently used in lipidic delivery vehicles, including liposomes and LNPs. These carriers generally took the form of (a) complexes and emulsions (such as lipoplexes, lipopolyplexes and cationic nanoemulsions), whereby a nucleic acid is mixed with and / or adsorbed onto the surface of one or more lipids; and (b) nanoparticles (such as liposomes, micelles and LNPs), whereby nanosized particles encapsulate or entrap nucleic acids within the body of the particle.
Lipoplexes
541. Some of the earliest lipid-based formulations used to deliver nucleic acids (primarily for in vitro applications) were lipoplexes. A lipoplex generally refers to a self-assembled mixture of: (i) one or more lipids (with a net positive charge), which are usually present in excess; and (ii) anionic (negatively charged) nucleic acid molecules.
Liposomes
542. A liposome is a particle whereby the positively charged components of the cationic lipid(s) have formed electrostatic interactions with the negatively charged phosphate (-PR) groups of the nucleic acids, such that the nucleic acids are adsorbed onto the surface of and / or encapsulated within a liposomal vesicle.
543. Liposomes are generally regarded as spherical particulate vesicles comprising an aqueous lumen (core) surrounded by one or more layers of lipids. They may be unilamellar (having a single phospholipid bilayer enclosing its aqueous core) or multilamellar (having an onion-like structure with concentric phospholipid bilayers separated by layers of water) or have more complex physical structures.
544. This technology was developed in the late 1980s and early 1990s. By the EP565 Priority Date, liposomes had been used for the encapsulation and in vivo delivery of nucleic acids, including RNA and DNA.
545. The term 'liposome' was commonly employed to refer to either the empty vesicle or the vesicle along with its encapsulated payload.
Lipid nanoparticles (LNPs)
546. LNPs had been used to encapsulate nucleic acids, including self-amplifying mRNA and siRNA, by the EP565 Priority Date. LNPs typically contain the following four main components:
a) A cationic lipid: This component may be permanently cationic, but by the EP565 Priority Date, it was preferable to use an ionisable cationic lipid - in the latter case, the lipid will be protonated (i.e., become positively charged) at a pH which is lower than its pKa, but will be relatively neutral at physiological pH (approximately 7.4). In the case of a nucleic acid payload such as RNA, the positively charged cationic lipid (when protonated, in the case of an ionisable lipid) forms electrostatic interactions with the anionic (negatively charged) nucleic acid to entrap the payload within the LNP. Once the LNP formulation has been delivered to the cell via endocytosis, the cationic lipid is also primarily responsible for the release of the payload into the cytoplasm of the cell via the process of endosomal escape.
b) A non-cationic / neutral lipid: promotes fusion with the cell and endosomal membrane. Sometimes referred to as a helper lipid, it has a neutral or near neutral net charge and plays a key role in the overall stability of the LNP and endosomal escape. It is usually a phospholipid such as DSPC, DPPC, POPC, DOPE or SM, although many other non-cationic / neutral lipids have also been considered. The most commonly used phospholipid is DSPC.
c) A structural lipid: The structural lipid (which is also a non-cationic / neutral lipid) plays a role in enhancing the overall stability of the nanoparticle by regulating the rigidity of its membrane. The most common structural lipids used in LNPs are sterols, particularly cholesterol, which is hydrophobic with a rigid structure which helps to promote the stability of the LNP particles.
d) A PEGylated lipid: which consists of a polyethylene glycol (PEG) molecule conjugated to a long chain alkyl group. It serves to protect the LNP from opsonisation in the serum, reduce clearance time, and improve stability during storage. They are usually present on the surface of the LNP formulation to prevent particle aggregation (during formulation and upon in vivo administration), while also increasing circulation time. Varying the type and quantity of PEGylated lipid, for example by reference to lipid length and molar mass, can affect the overall size and encapsulation efficiency of the LNP formulation, which also has a knock-on effect on its biodistribution and pharmacokinetics. Excessive PEGylation can inhibit cellular uptake and interactions with serum proteins by rendering the particle too stable.
547. Co-formulation of the lipid components together with the nucleic acid leads to encapsulation of the nucleic acid.
Cationic nanoemulsions
548. Microfluidics techniques are used to create an oil in water emulsion. These can be modified to include a cationic lipid, which allows the nucleic acid to interface with the particles once they have been pre-formed to create a complex via electrostatic interactions. In this way, it was shown that RNA could be protected from nucleases.
549. Ten disputed areas were identified in the Summary of Disputed CGK provided to me. Of these, only two remained in issue by the time of closing submissions (I retain the lettering from the list of ten):
a) the skilled team's view as to whether, and if so to what extent, betacoronaviruses were considered a vaccine development target at the EP565 Priority Date; and
e) the skilled team's view as to the relevant factors for an antigen-specific immune response by a nucleic acid vaccine.
Issue (a): Whether betacoronaviruses were a vaccine target at the EP565 Priority Date
550. Pfizer/BioNTech contended that after the SARS-CoV and MERS-CoV outbreaks (described above in the Agreed CGK section), there was ongoing concern at the EP565 Priority Date as to the threat posed by SARS-CoV and MERS-CoV. They pointed to a number of publications showing that MERS-CoV (and to some extent, SARS-CoV) was the subject of coordinated efforts by international organisations to develop a vaccine. Professor Dougan exhibited a number of documents to his second report relating to these efforts:
i) A statement by WHO dated 3 September 2015 and entitled "WHO statement on the tenth meeting of the IHR Emergency Committee regarding MERS". This document warns of the significant risk for further outbreaks of MERS and states that the outbreak in the Republic of Korea "demonstrated that when the MERS virus appears in a new setting, there is great potential for widespread transmission and severe disruption to the health system and to society". The Committee explicitly advised "International collaboration to develop human and animal vaccines and therapeutics should be accelerated";
ii) A copy of an announcement from September 2015 by the International Vaccine Institute (IVI) entitled "IVI Initiates MERS Vaccine Research and Development". This document records that IVI had launched a project to accelerate research and development of vaccines against MERS-CoV, with financial support from Samsung;
iii) A document from WHO dated 8 - 9 September 2015 entitled "December 2015 - First list of top emerging diseases likely to cause major epidemics" announcing that after a panel of scientists and public health experts convened by WHO had met in Geneva, the initial list of disease priorities determined as needing urgent R&D attention included MERS and SARS coronavirus diseases;
iv) A document from the Global Research Collaboration for Infectious Disease Preparedness (GLOPID-R) dated October 2015 and entitled "Overview of Funded Research on MERS-CoV". This document provides a helpful summary of vaccines in development at the time, including both human and camel vaccine candidates. Moderna said that this paper reinforces the idea that to the limited extent that there was any interest to do anything with a betacoronavirus vaccine, tried and tested approaches were preferred;
v) A document entitled "A Roadmap for Research and Product Development against Middle East Respiratory Syndrome-Coronavirus (MERS-CoV)" which resulted from a consultation held by WHO in December 2015. This document identifies the development and licensing of human and camel vaccines as a priority area; and
vi) A document published by WHO in May 2016 entitled "An R&D Blueprint for Action to Prevent Epidemics: Plan of Action", which was based on the roadmap listed above. One of the diseases listed to be urgently addressed was "Highly pathogenic emerging coronaviruses relevant to humans (MERS Co-V & SARS)".
551. Moderna contended that MERS/SARS were just two diseases in a list of eight pathogens identified by WHO as top emerging pathogens. It said there was no reason to single them out. In his oral evidence Dr Ulmer stated that if one compares the number of cases and deaths attributed to the viruses in the WHO list, SARS and MERS have much lower numbers than some of the other diseases and therefore the need for SARS and MERS seemed to be much lower.
552. Dr Ulmer also explained in his oral evidence that the lack of interest in coronavirus vaccines was not just a financial consideration, but also down to the fact that teams would have to divert time and resources from the development of one vaccine (where Moderna submitted there might be greater need) to the development of a coronavirus vaccine.
553. Professor Dougan also referred to two papers:
i) Excler JL et al., Toward Developing a Preventative MERS-CoV Vaccine - Report from a Workshop Organized by the Saudi Arabia Ministry of Health and the International Vaccine Institute, Riyadh, Saudi Arabi, November 14-15, 2015, Emerging Infectious Diseases 2016; 22(8): 1-7 ("Excler 2015"). This paper was authored by a group of experts convened by IVI and the Saudi government in November 2015. The paper states that MERS "remains a serious international public health threat within Saudi Arabia and internationally, as recently illustrated by an outbreak in South Korea with potential pandemic risk (1-7). A vaccine (or vaccines) targeting the MERS coronavirus (MERS-CoV), which causes the disease, will be a critical component of future public health prevention measures (8-10)." The paper specifically calls for the acceleration of development of vaccines and says priority should be given to platforms with an established safety track record in humans due to the urgency of the situation. Moderna emphasises that this paper does not refer to mRNA vaccines; and
ii) Modjarrad, L. et al., A roadmap for MERS-CoV research and product development: report from a World Health Organization consultation, Nat Med 2016; 22(7): 701-1005 ("Modjarrad 2016"). This paper reports on the consultation of leading experts in December 2015 convened by WHO. They were tasked with developing a roadmap for MERS-CoV research and development. The parties agreed that this paper would be CGK at least to a virologist with a specific interest in coronaviruses. It contains a helpful summary of MERS-CoV vaccine candidates in development (see figure below), showing that at least 13 MERS-CoV vaccine candidates were being developed at the time, and states that the global will to develop a coronavirus vaccine had "gained renewed momentum in the face of the current MERS-CoV outbreak". The roadmap itself (also agreed to be CGK as it was annexed to Modjarrad 2016), which was published in May 2016, states that there is "an urgent need for applied research and product development in order to better characterise epidemiology, diagnose, treat and prevent transmission and disease related to MERS-CoV." Moderna contended that again, there is no mention of RNA vaccines.

Table 2 from Modjarrad 2016.
554. Pfizer/BioNTech relied on the following paragraph from Modjarrad 2016 in their written closing:
The emergence and persistence of MERS-CoV as a cause of severe respiratory disease 10 years after the outbreak of severe acute respiratory syndrome coronavirus (SARS-CoV) highlights the need for the rapid development of effective interventions against highly pathogenic human coronaviruses. As MERS-CoV grows in global importance - causing disease and death in more than 1,700 and 600 people, respectively, across 27 countries - research and development (R&D) efforts to design diagnostic, prophylactic and therapeutic products are gaining momentum.
555. Modjarrad 2016 also provides evidence of a growing pipeline of MERS-CoV vaccines in development at the time. Table 2 (above) indicates that several vaccine platforms were at the stage of neutralising antibodies ("immunogenicity") and showing protective effect in an animal model ("efficacy"). The coronavirus virologist would therefore have been made aware, if they were not already aware, of these developments from the Modjarrad 2016 paper.
556. In their written closing, Pfizer/BioNTech pointed to the introduction to the Nidovirus symposium in 2014, written by Dr Sola and Dr Luis Enjuanes on behalf of the scientific and organising committees, as reflecting the position of Prof Dougan. The introduction included the following:
...The World Health Organization has issued a global alert for MERS-CoV (http://www.who.int/csr/disease/coronavirus_infections/en/). CoVs are emerging and re-emerging viruses that can mutate to extend their host range, potentially causing devastating pandemics... The rapid development of novel vaccines and antiviral therapies against CoVs that cause acute respiratory syndromes like SARS and MERS is an important priority for governments and public health agencies worldwide.
557. They also pointed to the transcript of Dr Ulmer's speech at the 4th International Conference on Vaccines and Vaccination in September 2014, where he called out SARS-CoV and MERS-CoV as being "emerging dangerous infection agents" which would benefit from a rapid response vaccine. I have addressed this above in my assessment of Dr Ulmer as a witness.
558. Pfizer/BioNTech relied on a number of other documents (several of which post-date the EP565 Priority Date) which were introduced to the case during the cross-examination of Moderna's experts:
i) In 2016 WHO launched a public consultation seeking proposals for new platform technologies to address WHO's priority diseases. Eight proposals were submitted relating to vaccines and Pfizer/BioNTech pointed to GSK's bid and a joint proposal from Janssen and the Jenner Institute. These bids were recorded in the agenda for a Coalition for Epidemic Preparedness Innovations (CEPI) interim board meeting held in August 2015;
ii) A newsletter from The Jenner Institute published in Autumn 2016 stated that the Jenner MERS-CoV vaccine would be entering into a phase I trial in the UK. It also states that "MERS has been chosen as a very high priority disease for accelerated vaccine development by the WHO, international vaccine experts (polled by Science magazine, 1 January 2016) and by members of the UK Vaccines Research and Development Network";
iii) CEPI was established after the World Economic Forum meeting in Davos in January 2016. It convened an expert taskforce to establish priority pathogens for CEPI to fund vaccine candidates against. In the draft Business Plan published by CEPI in August 2016 (and in the approved form published in October 2016), the taskforce designated coronaviruses (SARS-CoV and MERS-CoV) as Group 1 priority pathogens being "first choices for immediate funding";
iv) CEPI published its initial target diseases in a paper in early 2017, they were MERS, Lassa and Nipah;
v) Pfizer/BioNTech relied on publications from Moderna announcing their launch of a new venture "Valera" in January 2015 to advance mRNA vaccines and mRNA-based passive immunity therapies, and announcing the appointment of Dr Michael Watson (who was on the CEPI taskforce) as President of Valera in April 2016;
vi) Pfizer/BioNTech submitted in their written closing that it is notable that [0009] of EP565 which states "The outbreaks of MERS-CoV have raised serious concerns world-wide, reinforcing the importance of developing effective and safe vaccine candidates against MERS-CoV" had been taken from Du and Jiang, Middle East respiratory syndrome: current status and future prospects for vaccine development", Expert Opi. Biol Ther. 2015; 15(11): 1647-51 ("Du and Jiang"); and
vii) Pfizer/BioNTech also pointed to a paper published in January 2015, Regla-Nava et al, Severe Acute Respiratory Syndrome Coronaviruses with Mutations in the E Protein Are Attenuated and Promising Vaccine Candidates, J Virol 2015; 89(7): 3870-3887. Pfizer/BioNTech relied on the following quote in their written closing:
Human coronaviruses are important zoonotic pathogens. SARS-CoV caused a worldwide epidemic infecting more than 8,000 people with a mortality of around 10%. Therefore, understanding the virulence mechanisms of this pathogen and developing efficacious vaccines are of high importance to prevent epidemics from this and other human coronaviruses.
When Counsel for Pfizer/BioNTech put this to Dr Sola, she accepted that it was a fair assessment.
559. These materials paint an extremely clear picture that coronaviruses were widely regarded as important vaccine targets, especially for SARS and MERS, and that it was recognised that there was a high risk of outbreaks that would need to be addressed quickly. These matters were widely known among the international vaccine community, both in the public and private sector, and were CGK. They were well known both to people with a narrow interest in coronaviruses and to those in the international vaccine community with an interest in identifying more broadly those diseases where vaccine development resources might best be deployed - I refer back to what I have said above about the EP565 skilled team.
560. I acknowledge that some of Pfizer/BioNTech's materials are post-priority and for present purposes I agree with Moderna that materials that only came into the case in time for cross-examination are entitled to much less weight, if any, unless their force is accepted by the witness to whom they are put. But those points only chip away at the edges of a really substantial body of materials which prove Pfizer/BioNTech's points. In a sense Pfizer/BioNTech could have made good this case from Modjarrad 2016 alone, which was accepted to be CGK (albeit Moderna said, but I disagree, only to those familiar with coronaviruses).
561. I agree with Moderna's point that the materials referred to above only show the development of coronavirus vaccines with established platforms, and that although they do include nucleic acid vaccines they do not extend to RNA vaccines. It would be the state of the CGK, therefore, that existing efforts on coronavirus vaccines were in fact directed to established platforms, but that does not mean that it was CGK that they had to be, or needed to continue to be.
Geall 2012
562. As I have just touched on, somewhat rolled into disputed issue (a) was Moderna's argument that insofar as there was CGK of coronavirus vaccines, it was only understood to be ongoing in relation to established platforms. To answer this point (and for other purposes) Pfizer/BioNTech relied on Geall 2012, an article publishing work by the Novartis group. It is co-authored by Dr Ulmer.
563. The parties agreed that Geall 2012 was a CGK reference.
564. Geall 2012 relates to an sa mRNA RSV vaccine encoding the RSV-F protein, encapsulated in LNPs with components in ratios that Pfizer/BioNTech say fall within claim 1 of EP565. The authors state that they used LNPs which had been shown to be effective for the delivery of siRNA. It provides in vivo data demonstrating that sa mRNA encapsulated in LNPs administered in low doses can achieve high potency when compared to naked sa mRNA and sa mRNA encapsulated in viral replicon particles (VRPs).
565. The authors refer to the application of their technology as a platform: the "SAM vaccine platform". They state that the platform has the potential to "address multiple disease targets" and that the "unique nucleic acid vaccine technology could enable a new generation of potent, versatile, and easily produced vaccines to address the health challenges of the 21st century." Pfizer/BioNTech pointed to this as an example of how Novartis promoted its SAM vaccine platform as suitable for adaptation to a range of pathogens. Pfizer/BioNTech said this correlates with Dr Ulmer's evidence that the Geall team received numerous requests to collaborate in the development of vaccines for many different disease targets after the publication of results from Novartis' saRNA RSV vaccine project in 2012.
566. I agree with Pfizer/BioNTech on the significance of Geall 2012. Being CGK, it would lead to the skilled team thinking that there was unlikely to be difficulty applying LNPs in the context of mRNA (as opposed to siRNA) and, more importantly in response to Moderna's point, that LNPs could reasonably be tried for delivering mRNA for vaccine purposes for a range of pathogens: that is what "platform" implies.
567. Other somewhat similar work relating to influenza was reported in a paper by Hekele et al in 2013 but it was not agreed to be CGK and is not necessary to my conclusions anyway.
Issue (e): The skilled team's view as to the relevant factors for an antigen-specific immune response by a nucleic acid vaccine
568. Moderna's position was that there are several factors which affect the efficacy of a nucleic acid vaccine. Moderna's closing skeleton explained that delivery of an LNP formulation does not always lead to expression, and expression does not always equal immunogenicity. Dr Ulmer reiterated this during cross-examination and further explained that "you cannot assume that because one antigen or protein is expressed that another one under the same conditions will express". Relatedly, during cross-examination, Dr Sola stated that the only example providing information about the platform in WO674 related to influenza, but the antigen for influenza is very different to the spike protein of betacoronaviruses, meaning one could not predict that the same platform would produce a protective effect if used for betacoronaviruses. Moderna characterised Pfizer/BioNTech's position as simply requiring delivery of the nucleic acid in order to get expression. I think this is an oversimplification of the position of the experts called by Pfizer/BioNTech.
569. Counsel for Pfizer/BioNTech put the paragraphs in Dr Ulmer's report relating to the matters which he said can affect the efficacy of a nucleic acid vaccine to him in cross-examination:
Q. Looking at these factors, the nucleic acid vaccinologist would know, would they not, that their prospects of success are increased by selecting an antigen that is known to produce neutralising antibodies?
A. That would be encouraging, yes.
Q. And by selecting a delivery vehicle that is known to be effective in delivering nucleic acids into cells, leading to expression?
A. Yes.
Q. And by avoiding the use of DNA?
A. Yes.
570. In their closing skeleton, Pfizer/BioNTech pointed to Dr Sola's evidence that the S protein was known to be the main inducer of neutralising antibodies following infection by MERS or SARS, and that sufficiently high levels of neutralising antibodies were associated with protection. In her oral evidence, Dr Sola explained that, due to the conformational nature of most of the neutralising epitopes, one needs enough of the protein, properly folded, to induce neutralising antibodies. In answer to this, Pfizer/BioNTech pointed to Dr Ulmer's written evidence, which noted that one of the known advantages of nucleic acid vaccines in 2015 was their effectiveness. Dr Ulmer also agreed in cross-examination that he would expect any vaccinologist working on a vaccine project to make sure they were informed about other vaccine candidates for the target pathogen. Pfizer/BioNTech submitted that the skilled team would know from this information that in relation to coronaviruses a) there were several vaccine candidates in development, most of which used the S protein or a subunit as the target antigen, b) many of the candidates were nucleic acid-based and c) the nucleic acid vaccines encoding the S protein had been shown to elicit neutralising antibodies and several candidates had shown protective effect in mice and/or primates. The skilled team would recognise that the S protein was being properly folded, processed and glycosylated in order to cause this protective effect. I accept this submission.
571. As a matter of CGK, I also find (and this was not really disputed) that it was CGK in connection with nucleic acid vaccines that an antigen that produced neutralising antibodies ought to be chosen, that an effective delivery vehicle was necessary, and that DNA was best avoided.
572. The importance of the S protein and that it produced neutralising antibodies and protective effects in animal models was also CGK to those familiar with coronaviruses. The skilled person would understand as a matter of CGK that the facts that neutralising antibodies were produced and a protective effect achieved, meant that the S protein had indeed been adequately processed, folded and glycosylated.
573. It was also, however, CGK, that the requirements for a nucleic acid vaccine to be effective (to be delivered into the cell, to be expressed and folded properly) could not be guaranteed to work in any given situation and would ultimately need to be tested.
574. EP565 is over 500 pages long and repetitive in places; it would be impractical and unhelpful to try to summarise it all. The arguments focused on some significant aspects of the specification and I will do the same.
575. The title of EP565 is "Respiratory Virus Vaccines". Whilst the claims are directed to betacoronavirus vaccines, the description is much broader. The "Background" section describes the following viruses: human metapneumovirus (hMPV), parainfluenza virus type 3 (PIV3), respiratory syncytial virus (RSV) and measles virus (MeV), along with Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome (SARS). EP565 applies the same mRNA-LNP vaccine approach to each of these viruses.
576. When introducing MERS-CoV in the "Background" section at [0009], it explains:
As of July 2015, MERS-CoV cases have been reported in over 21 countries. The outbreaks of MERS-CoV have raised serious concerns world-wide, reinforcing the importance of developing effective and safe vaccine candidates against MERC-CoV.
577. When introducing SARS in [0010], it explains:
Severe acute respiratory syndrome (SARS) emerged in China in 2002 and spread to other countries before brought under control. Because of a concern for reemergence or a deliberate release of the SARS coronavirus, vaccine development was initiated.
578. The statements in [0009] and [0010], especially the former, are strongly in line with Pfizer/BioNTech's position on the CGK of coronaviruses as vaccine targets, and contrary to Moderna's position. It is a further piquant detail that [0009] appears to be based on a contemporary publication (Du and Jiang) as I have touched on above.
579. Statements of this kind by a patentee are, Counsel for Moderna accepted, part of the materials that the Court can take into account in assessing the state of the art/CGK. At the same time, it is well established that they do not irretrievably bind the patentee and are not conclusive. Other evidence might show that the patentee was wrong in making the statement. In the present case, as I say, these paragraphs strongly support Pfizer/BioNTech and Moderna is nowhere near to showing that the patentee was wrong. I would, though, have reached exactly the same conclusion if they had not been there, because of the strong objective evidence in the contemporary literature.
580. [0011] refers to DNA vaccination as a technique used, but it states that the technique comes with potential problems such as insertional mutagenesis.
581. Under the "Summary" heading, [0014] states:
Based on the disclosure that is contained herein, the present invention provides a betacoronavirus (BetaCoV) messenger RNA (mRNA) vaccine comprising at least one mRNA polynucleotide having an open reading frame encoding at least one BetaCoV antigenic polypeptide; wherein the at least one BetaCoV antigenic polypeptide is (a) a spike (S) protein or immunogenic fragment thereof, or (b) an S1 subunit or an S2 subunit of S protein or an immunogenic fragment thereof; wherein the BetaCoV vaccine is formulated in a lipid nanoparticle, wherein the lipid nanoparticle comprises 40-60% cationic lipid, 5-15% non-cationic lipid, 1-2% PEG lipid, and 30-50% cholesterol.
582. The "Detailed Description" section starts at [0017] and in [0019] it states:
Provided herein are ribonucleic acid (RNA) vaccines that build on the knowledge that messenger RNA (mRNA) can safely direct the body's cellular machinery to produce nearly any protein of interest, from native proteins to antibodies and other entirely novel protein constructs that can have therapeutic activity inside and outside of cells. The mRNA vaccines of the present disclosure may be used to induce a balanced immune response against BetaCoV (e.g., MERS-CoV, SARS-CoV, HCoV-OC43, HCoV-229E, HCoV-NL63, HCoV-NL, HCoV-NH and/or HCoV-HKU1), comprising both cellular and humoral immunity, without risking the possibility of insertional mutagenesis, for example.
583. There is some teaching about the advantages of mRNA vaccines at [0020]:
The RNA (i.e.., mRNA) vaccines may be utilized in various settings depending on the prevalence of the infection or the degree or level of unmet medical need. The RNA (i.e.., mRNA) vaccines have superior properties in that they produce much larger antibody titers and produce responses earlier than commercially available anti-viral therapeutic treatments. While not wishing to be bound by theory, it is believed that the RNA (i.e.., mRNA) vaccines, as mRNA polynucleotides, are better designed to produce the appropriate protein conformation upon translation as the RNA (i.e.., mRNA) vaccines co-opt natural cellular machinery. Unlike traditional vaccines, which are manufactured ex vivo and may trigger unwanted cellular responses, RNA (i.e.., mRNA) vaccines are presented to the cellular system in a more native fashion.
584. The "BetaCoV" section begins at [0030]. It explains that the antigenic polypeptide in the invention is a betacoronavirus structural protein, and in some embodiments the betacoronavirus structural protein is a spike protein. In [0031] it explains that vaccines with RNA polynucleotides encoding structural proteins are believed to have preferred immunogenic activity because of their surface expression properties, so they may be the most suitable as vaccines. It was common ground between the experts that the S protein, or immunogenic parts thereof, was the leading candidate for an immunogen for a betaCoV vaccine and it was known to induce neutralising antibodies.
585. mRNA is defined in [0175] under the heading "Nucleic Acids/Polynucleotides":
Polynucleotides of the present invention function as messenger RNA (mRNA). "Messenger RNA" (mRNA) refers to any polynucleotide that encodes a (at least one) polypeptide (a naturally-occurring, non-naturally-occurring, or modified polymer of amino acids) and can be translated to produce the encoded polypeptide in vitro, in vivo, in situ or ex vivo. The skilled artisan will appreciate that, except where otherwise noted, polynucleotide sequences set forth in the instant application will recite "T"s in a representative DNA sequence but where the sequence represents RNA (e.g., mRNA), the "T"s would be substituted for "U"s. Thus, any of the RNA polynucleotides encoded by a DNA identified by a particular sequence identification number may also comprise the corresponding RNA (e.g., mRNA) sequence encoded by the DNA, where each "T" of the DNA sequence is substituted with "U".
586. At [0285] a section on "Nanoparticle Formulations" begins. It explains that the invention relates to vaccines formulated in a lipid nanoparticle. [0286] states:
A lipid nanoparticle formulation may be influenced by, but not limited to, the selection of the cationic lipid component, the degree of cationic lipid saturation, the nature of the PEGylation, ratio of all components and biophysical parameters such as size. In one example by Semple et al. (Nature Biotech. 2010 28:172-176), the lipid nanoparticle formulation is composed of 57.1% cationic lipid, 7.1% dipalmioylphosphatidylcholine, 34.3% cholesterol, and 1.4% PEGc-DMA. As another example, changing the composition of the cationic lipid can more effectively deliver siRNA to various antigen presenting cells (Basha et al. Mol Ther. 201119:2186-2200).
587. Much of the rest of the description was largely irrelevant to the parties' submissions. However, both parties placed emphasis on specific examples.
588. There are 26 examples in EP565 and they can be grouped as follows:
i) Examples 1 to 11: These examples set out a number of established methods for the manufacture, formulation and characterisation/quantification of polynucleotides for use in RNA-based vaccines. Nothing turns on these examples.
ii) Examples 12 to 19: These are all reference examples relating to hMPV (examples 12 to 16), PIV3 (examples 17 to 18), or both (example 19). Professor Dougan, Dr Ulmer and Dr Sola all stated that at least some aspects of this section would be of interest to the NAV and Coronavirus Virologist.
iii) Examples 20 to 24: These are the betacoronavirus examples and it was common ground between the parties that the skilled team would be most interested in these examples.
iv) Examples 25 to 26: These are study protocols for an immunogenicity study and a challenge study in mice for a MeV mRNA vaccine. No data is provided and nothing turns on them.
The reference examples (examples 12 to 19)
589. It is not necessary to reproduce the detail of each example. I shall focus on the significant passages to which I was referred.
590. Example 13 is a challenge study in rats designed to test the efficacy of hMPV vaccine candidates. No data is provided in this example but [0453] specifies the LNP formulation that may be used:
In experiments where a lipid nanoparticle (LNP) formulation is used, the formulation may include a cationic lipid, non-cationic lipid, PEG lipid and structural lipid in the ratios 50:10:1.5:38.5. The cationic lipid is DLin-KC2-DMA (50 mol%) or DLin-MC3-DMA (50 mol%), the non-cationic lipid is DSPC (10 mol%), the PEG lipid is PEG-DOMG (1.5 mol%) and the structural lipid is cholesterol (38.5 mol%), for example.
591. There was a dispute between the parties as to the teaching of the Patent with respect to whether the betacoronavirus examples used an LNP as claimed in EP565 or not. It was Dr Ulmer's evidence that it appeared that the LNP formulation in paragraph [0453] was specified for Example 13 and was used in each of the hMPV reference examples. Pfizer/BioNTech submit that the wording of [0453] (emphasising "where a lipid nanoparticle (LNP) is used" and "the formulation may include") does not specify any formulation for use either in Example 13 or in any other example.
592. The same wording appears in [0467] under Example 18 and [0476] under Example 20. Moderna relied on this (along with a number of references from the description, including [0014], [0030], [0484] and [0486]) as evidence that the skilled person would understand that all the examples using an LNP were formulated using the ratio set out in this LNP formulation. Moderna also pointed to Professor Alabi's statement that the LNP formulation of claim 1 was "entirely standard". These were really points directed to the added matter arguments and I return to them (in particular Prof Alabi's statement) there. In my view the skilled person would think that it was possible that LNPs as claimed were used and that it would make sense, but would not be certain of it.
593. Pfizer/BioNTech referred to Example 16 in their written closing. Example 16 is a challenge study to test the efficacy of hMPV vaccines in cotton rats. In his first report Dr Ulmer said as follows when analysing Figs 9A and 9B of Example 16:
The 10 μg dose of mRNA vaccine encoding hMPV F protein shows substantial protection with undetectable levels of virus in the lungs (the reading is below the dotted line), and reduced levels of virus in the nose, indicating some protective effect. The 2 μg dose and FI vaccine show some reduction of the virus in the lung, but not in the nose.
594. During cross-examination, Dr Ulmer confirmed that both doses show some protective effect. Pfizer/BioNTech say this indicates the degree of protective effect needed to fall within claim 10 and shows the low level of effect against which I should judge the skilled team's expectation of success for obviousness. I do not agree with this, at least not to the extent that Pfizer/BioNTech was arguing that the prospect of obtaining merely this level of effect would motivate a skilled team to set out on a vaccine development project, but it does not matter because Pfizer/BioNTech's obviousness case was based around a skilled team setting out to develop a functioning, effective vaccine in the normal sense, not at this lower level.
The betacoronavirus examples (examples 20 to 24)
595. Example 20 is a method for testing the immunogenicity in rabbits of candidate betacoronavirus mRNA vaccines. As mentioned above, it provides an example LNP formulation.
596. Example 21 is a study design of a challenge model to test the efficacy of candidate betacoronavirus mRNA vaccines encoding the S, S1 or S2 protein in rabbits.
597. Example 22 describes a microneutralization assay to assess neutralising antibodies against a betacoronavirus.
598. No data are provided in Examples 20 to 22.
599. The parties focused particularly on Examples 23 and 24. Example 23 tests the immunogenicity in mice of candidate MERS-CoV vaccines. The example is described in [0480] to [0482] as follows:
[0480] The instant study was designed to test the immunogenicity in mice of candidate MERS-CoV vaccines comprising a mRNA polynucleotide encoding the full-length Spike (S) protein, or the S2 subunit (S2) of the Spike protein obtained from MERS-CoV.
[0481] Mice were vaccinated with a 10 mg dose of MERS-Co V mRNA vaccine encoding either the full-length MERS-CoV Spike (S) protein, or the S2 subunit (S2) of the Spike protein on days 0 and 21. Sera were collected from each mice on days 0, 21, 42, and 56. Individual bleeds were tested for anti-S, anti-S2 activity via a virus neutralization assay from all four time points.
[0482] As shown in Fig. 17, the MERS-CoV vaccine encoding the full-length S protein induced strong immune response after the boost dose on day 21. Further, full-length S protein vaccine generated much higher neutralizing antibody titers as compared to S2 alone (Fig. 18).
600. The results are shown in Figures 17 and 18. I do not think it necessary to reproduce the figures given the eventual state of the evidence.
601. There was a modest dispute between the parties in relation to the experiment and the data. However, both parties agreed that the data show that higher levels of neutralising antibodies are generated by the vaccine which encodes the full length of the S protein, compared to the vaccine encoding the S2 subunit. It also shows that the vaccine encoding the full-length S protein had generated neutralising antibodies at day 21 and generation increases with time.
602. In his first report, Prof Dougan gave eight reasons why, in his opinion, the data in Figures 17 and 18 would have a limited impression on the vaccinologist. The reasons include that it is not clear how many mice were tested, that the error bars are large in some instances and that it is not clear by which route(s) the vaccine was administered. While there may have been something in each of these criticisms, and other similar ones made about Example 24 (see below), they do not undermine the overall conclusions that could be drawn, as Prof Dougan accepted.
603. Example 24 is a challenge study which tests the immunogenicity of candidate MERS-CoV mRNA vaccines encoding the full-length S protein in rabbits compared to unvaccinated rabbits. It is described in paragraphs [0483] to [0486]:
[0483] The instant study was designed to test the immunogenicity of candidate MERS-CoV mRNA vaccines encoding the full-length Spike (S) protein. The New Zealand white rabbits used in this study weighed about 4-5 kg. The rabbits were divided into three groups (Group 1a, Group 1b, and Group 2, n=8). Rabbits in Group 1a were immunized intramuscularly (IM) with one 20 mg dose of the MERS-CoV mRNA vaccine encoding the full-length Spike protein on day 0. Rabbits in Group 1b were immunized intramuscularly (IM) with one 20 mg dose of the MERS-CoV mRNA vaccine encoding the full-length Spike protein on day 0, and again on day 21 (booster dose). Group 2 received placebo (PBS). The immunized rabbits were then challenged and samples were collected 4 days after challenge. The viral loads in the lungs, bronchoalveolar lavage (Bal), nose, and throat of the rabbits were determined, e.g., via quantitative PCR. Replicating virus in the lung tissues of the rabbits were also detected. Lung histopathology were evaluated and the neutralizing antibody titers in serum samples of the rabbits were determined.
[0484] Two 20 mg doses of MERS-CoV mRNA vaccine resulted in a 3 log reduction of viral load in the nose and led to complete protection in the throat of the New Zealand white rabbits (Fig. 19A). Two 20 mg doses of MERS-CoV mRNA vaccine also resulted in a 4 log reduction of viral load in the BAL of the New Zealand white rabbits (Fig. 19B). One 20mg dose of MERS-Co V mRNA vaccine resulted in a 2 log reduction of viral load, while two 20 mg doses of MERS-CoV mRNA vaccine resulted in an over 4 log reduction of viral load in the lungs of the New Zealand white rabbits (Fig. 19C).
[0485] Quantitative PCR results show that two 20 mg doses of MERS-CoV mRNA vaccine reduced over 99% (2 log) of viruses in the lungs of New Zealand white rabbits (Fig. 20A). No replicating virus were detected in the lungs (Fig. 20B).
[0486] Further, as shown in Fig. 21, two 20 mg doses of MERS-CoV mRNA vaccine induced significant amount of neutralizing antibodies against MERS-CoV (EC50 between 500-1000). The MERS-CoV mRNA vaccine induced antibody titer is 3-5 fold better than any other vaccines tested in the same model.
604. The results are shown in Figures 19-21. Again, I do not think it is necessary to reproduce them.
605. Fig. 19A shows that vaccination reduced the viral load in the throat. In the nose, vaccination reduced viral load, particularly after two doses.
606. Fig. 19B provides lung data and shows that two doses appeared to be more effective in reducing viral load than one dose.
607. Fig. 20A shows viral load in the lungs of the immunised groups. It can be seen that the RNA load in the placebo group (Group 2) is larger than both Group 1a and Group 1b. Group 1b (2 doses) shows a larger reduction than Group 1a (1 dose). This Figure correlates to Fig. 20B which shows the presence of replicating virus (TCID50) in the lungs. Both of the vaccinated groups show reduction in infectious virus.
608. Fig. 21 shows neutralising antibody titre data in immunised rabbits. The first immunisation provides an increase in neutralising antibodies, and the boost dose in Group 1b causes a large increase in neutralising antibodies. Titres increase for both groups post-challenge.
609. Pfizer/BioNTech took issue with the statement in [0486] that the titre shown in Fig. 21 was "3-5 fold better than any other vaccines tested in the same model", as no other vaccines are identified.
610. In their closing skeleton, Pfizer/BioNTech stated that Examples 23 and 24 have the following deficiencies:
i) They do not compare the mRNA vaccines with any other vaccines;
ii) They do not examine the cellular immune response;
iii) They do not compare delivery vehicles;
iv) They do not contain data on whether the immune response had a Th1 or Th2 bias or examine immunopathology; and
v) They do not specify whether the mRNA was chemically modified or not, or compare chemically modified mRNA with unmodified mRNA.
611. Again, these points may have some minor force but do not undermine the overall picture that there was a positive and useful effect. It was not submitted by Pfizer/BioNTech that they constituted or even supported any concrete invalidity attack. They were really just chipping away at EP565's general merits by pointing out that it does not meet the highest standards of experimental science, which is not what is required of a patent specification. So they were rather a waste of time.
612. The claims in issue are claims 1, 2, 6, 7 and 10 (as unconditionally amended) and claims 1A, 1B, 1C, 1D, 4, 5 and 8 as conditionally proposed to be amended. The real issues in the case can all be determined by reference to claims 1 and 10 as unconditionally amended, so I will just set them out here. I deal with all the other claims in a section at the end of this judgment.
613. Claim 1 is:
A. A betacoronavirus (BetaCoV) messenger RNA (mRNA) vaccine comprising
B. at least one mRNA polynucleotide having an open reading frame encoding at least one BetaCoV antigenic polypeptide;
C. wherein the at least one BetaCoV antigenic polypeptide is (a) a spike (S) protein or immunogenic fragment thereof, or (b) an S1 subunit or an S2 subunit of S protein or an immunogenic fragment thereof;
D. wherein the BetaCoV vaccine is formulated in a lipid nanoparticle, wherein the lipid nanoparticle comprises 40-60% cationic lipid, 5-15% non-cationic lipid, 1-2% PEG lipid, and 30-50% cholesterol.
614. Claim 10 is:
The vaccine of any of claims 1-9 for use in a method of preventing and/or treating a BetaCoV disease in a subject.
615. Two points of claim interpretation were flagged in the parties' written submissions.
616. The first was whether the claims excluded self-amplifying mRNA. This was only relevant to issues which fell away by the end of the trial so I do not need to decide it.
617. The second was the meaning of "vaccine", and the additional requirement of claim 10. If there ever was a dispute it dwindled away, and Pfizer/BioNTech agreed that:
i) Dr Sola's definition of vaccine as a composition which elicits an antigen-specific immune response was appropriate.
ii) For the purposes of claim 10, the vaccine needed to have a protective effect against betaCoV disease, typically demonstrated in a suitable animal model. Again, this was Dr Sola's point of view.
618. Pfizer/BioNTech was concerned to point out that for a vaccine to fall within claim 10 it did not need to avoid VAED or other side effects. I agree with this, but Pfizer/BioNTech raised it because of a concern that Moderna would make the avoidance of side effects part of the obviousness case, which did not happen.
619. WO674 is an international patent application filed on 23 April 2015 and published on 29 October 2015. The applicant is Moderna Therapeutics, Inc. The title is "Nucleic Acid Vaccines".
620. The "Field of the Invention" is described as follows:
The invention relates to compositions, methods, processes, kits and devices for the selection, design, preparation, manufacture, formulation, and/or use of vaccines, specifically nucleic acid vaccines (NAVs). In particular, the invention relates to compositions, methods, processes, kits and devices for the selection, design, preparation, manufacture, formulation, and/or use of ribonucleic acid (RNA) vaccines, e.g., mRNA vaccines.
621. The "Background" section describes typical vaccines and the obstacles that arise in the development of traditional vaccines. One of the obstacles described is the "constant evolution of most infectious agents, such as viruses and bacteria. Viruses often mutate their surface proteins to generate new antigens which can help them skipping the active immune system that has been immunized by vaccines containing the viruses."
622. The "Summary of the Invention" section stretches from page 2 to page 15 and describes various embodiments of the invention. A molar ratio for the LNP carrier is provided for "some aspects" of the invention at p318-34 to p41-7:
Aspects of the invention provide nucleic acid vaccines (NAVs) comprising one or more RNA polynucleotides having an open reading frame encoding a first antigenic polypeptide, formulated within a cationic lipid nanoparticle. Some aspects provide NAVs comprising one or more RNA polynucleotides having an open reading frame encoding a first antigenic polypeptide, formulated in a carrier having a molar ratio of about 20-60% cationic lipid: 5-25% non-cationic lipid: 25-55% sterol; and 0.5-15% PEG-modified lipid.
In some embodiments, the cationic lipid nanoparticle comprises a cationic lipid, a PEG-modified lipid, a sterol and a non-cationic lipid. In some embodiments, the cationic lipid is selected from the group consisting of 2,2-dilinoleyl-4-dimethylaminoethyl-[l ,3]-dioxolane (DLin-KC2-DMA), dilinoleyl-methyl-4-dimethylaminobutyrate (DLin-MC3-DMA), and di((Z)-non-2-en-1-yl) 9-((4-(dimethylamino)butanoyl)oxy)heptadecanedioate (L319). In some embodiments, the cationic lipid nanoparticle has a molar ratio of about 20-60% cationic lipid: about 5-25% non-cationic lipid: about 25-55% sterol; and about 0.5-15% PEG-modified lipid. In some embodiments, the cationic lipid nanoparticle comprises a molar ratio of about 50% cationic lipid, about 1.5% PEG-modified lipid, about 38.5% cholesterol and about 10% non-cationic lipid. In some embodiments, the cationic lipid nanoparticle comprises a molar ratio of about 55% cationic lipid, about 2.5% PEG lipid, about 32.5% cholesterol and about 10% non-cationic lipid. In some embodiments, the cationic lipid is an ionizable cationic lipid and the non-cationic lipid is a neutral lipid, and the sterol is a cholesterol. In some embodiments, the cationic lipid nanoparticle has a molar ratio of 50:38.5: 10: 1.5 of cationic lipid: cholesterol: PEG2000-DMG:DSPC.
623. It is stated that in any of the embodiments, the infectious agent is selected from a list of viruses which includes SARS and MERS (p429-510).
624. From p21 there is a section dedicated to nucleic acid vaccines. From p35 lists of infectious agents are provided including bacterial, viral (where SARS and MERS are listed), fungal, therapeutic settings, influenza and influenza antigens. After listing various Haemagglutinin Amino Acid Sequences, it goes on to describe MRSA, Dengue, Enterotoxic E.Coli, C. Difficile and Tuberculosis, and MERS.
625. No significance was placed on section III (Design, Synthesis and Quantification of NAV Polynucleotides), section IV (Modifications), section V (Pharmaceutical Vaccine Compositions), VI (Kits and Devices) or VII (Definitions).
Examples
626. WO674 contains 28 examples. Examples 15 and 21-28 provide results using influenza vaccines from in vivo studies on mice, non-human primates and ferrets.
627. At p59913-20 , in a paragraph which discusses the data presented in the Examples, it is stated that "it was discovered herein that chemically modified mRNA-LNP vaccines required a much lower effective mRNA dose than unmodified mRNA, i.e., tenfold less than unmodified mRNA."
628. Example 14 is called "Antigen Polynucleotides" and contains a table (Table 28) listing immunogens for viral targets. Various strains of Dengue are listed from rows 18 to 25, tuberculosis is listed in rows 59 to 61 and the MERS-CoV spike protein is listed in row 69. Moderna noted in its opening skeleton that 20 of the 82 antigens listed relate to influenzas.
629. Following Table 28, under the sub-heading "Pan-flu NAVs", it states:
Polynucleotides used in the studies herein which encode certain infectious agent antigens or variants thereof may be formulated in any of the formulations described herein including LNPs.
630. Example 15 is an influenza study in mice. It has two phases; firstly testing the immunogenicity of mRNA vaccines encoding an influenza antigen, and secondly testing the efficacy of candidate influenza vaccines in mice against a lethal challenge of influenza. The study design is set out in Table 31 and the LNP used is described on p87622-25:
The LNP formulation consisted of a cationic lipid, non-cationic lipid, PEG lipid and structural lipid in the ratios 50: 10: 1.5:38.5. The cationic lipid was DLin-KC2-DMA (50 mol%), the non-cationic lipid was DSPC (10 mol%), the PEG lipid was PEG-DOMG (1.5 mol%) and the structural lipid was cholesterol (38.5 mol%).
631. The results can be seen in tables 32 to 36 and in Fig.10. Table 32 compares the titres obtained using modified and unmodified mRNA, with modified mRNAs generally performing better than unmodified.
632. Example 16 is a study for MRSA vaccine candidates formulated in an LNP using the same composition as Example 15 but altering the cationic lipid to DLin-MC3-DMA (rather than DLin-KC2-DMA) for groups 8-13. It is noted on p866 that efficacy was not shown with either the RNA vaccine or the controls, suggesting "the model was not adequate for testing the constructs".
633. Example 17 is an immunogenicity study of a nucleic acid mRNA vaccine using a dengue virus antigen in mice. Data is provided and WO674 concludes on p888 that the "data suggests that a mRNA vaccine to a single dengue viral antigen can produce antibody in preliminary studies."
634. Example 20 is a MERS-CoV study for which no data are provided. After describing a passive immunity decoy approach, it states:
Another key vaccine for MERS-CoV is identified as an mRNA encoding MERS-CoV Spike Glucoprotein as an antigen. The protein sequence is given here (SEQ ID NO: 2275).
635. The protein sequence is then set out in full. "Another" just means in addition to the decoy approach.
636. Examples 21-28 are studies of mRNA vaccines encoding hemagglutinin antigens against influenza. They show that mRNA LNP formulated vaccines generated antibody responses in mice, ferrets and non-human primates. mRNA LNP formulated vaccines also showed efficacy in challenge studies in mice and ferrets. The use of LNPs is specified in examples 22, 23 and 25 to 28, with examples 22, 25, 26 and 28 specifically using either KC2 or MC3 LNP formulations.
637. Pardi 2015 is a paper entitled "Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes". It was published online on 8 August 2015.
638. The paper describes how modified non-sa mRNA encapsulated in LNPs were delivered to mice and the authors state in the Abstract that their "results demonstrate that LNPs are appropriate carriers for mRNA in vivo and have the potential to become valuable tools for delivering mRNA encoding therapeutic proteins."
639. In the Introduction, the authors list the hurdles that need to be overcome to use mRNA therapeutically, these are: poor translatability, lack of RNA stability, inefficient in vivo delivery and activation of innate immune sensors. They state that the efficient delivery of mRNA into target cells in vivo is a "major challenge". LNPs are identified as one of the most advanced technological platforms for delivering mRNA.
640. In the study, LNP-encapsulated, HPLC-purified, 1-methylpseudouridine-containing mRNA encoding firefly luciferase was delivered into cultured cells and mice. A variety of administrative routes including intravenous, subcutaneous and intradermal were used.
641. The authors state that the LNPs used "were similar in composition to those described previously [7,8], which contain ionizable cationic lipid/phosphatidylcholine/cholesterol/PEG-lipid (50:10:38.5:1.5 mol/mol)". References 7 and 8 are to two papers in which siRNA was delivered.
642. The Introduction concludes with the following sentence:
We have found that administration of mRNA-LNP complexes results in large amounts of protein production in vivo for varying lengths of time demonstrating that LNPs are suitable tools for highly efficient mRNA delivery.
643. The Discussion section begins on p349:
Nucleoside-modified mRNA has given new interest to the use of mRNA to express proteins for research and therapeutic purposes. mRNA does not require nuclear entry, it cannot induce insertional mutagenesis, and it results in both very rapid and highly controllable protein expression. Thus, the non-integrating and transient character of mRNA therapy is extremely well suited for using mRNA for expressing proteins and applying it for replacement therapies.
644. Later in the Discussion section, on p350, the authors state:
Expression pattern, dosing, duration of protein translation and the amount of produced protein are the key determinants for in vivo use of mRNA encoding therapeutic proteins. Intradermally and intranodally administered naked and protamine complexed mRNA vaccines have entered clinical trials (ClinicalTrials.gov identifiers: NCT01915524, NCT01817738 and NCT01684241) but there have been no human trials on systemic delivery of mRNA to replace or deliver a therapeutic protein. Different applications have varying requirements for dosing, duration of expression, intracellular versus extracellular function and site of activity. Moderate protein expression from the mRNA that also stimulates the innate immune system for a few days might be sufficient to induce immune responses but will not be successful for supplementing therapeutic proteins.
645. The authors also state that "[i]mportantly, very low doses (0.005 mg/kg) of mRNA-LNPs could be translated for several days following the tested delivery routes demonstrating the potential of these formulations for in vivo development."
646. In the Conclusion, the "major finding" is described to be "the outstanding ability of mRNA-LNPs for in vivo mRNA delivery." The authors go on to say:
To our knowledge, this is the first demonstration that a single injection of low dose (0.005-0.250 mg/kg) mRNA administered by various injection routes translated at high levels for up to 10 days depending on the dose and the site of the delivery.
647. Because, for reasons given below, I have reached the clear conclusion that EP565 is obvious over WO674 and because the issues on novelty and added matter are essentially ones of law, I am going to deal with them only briefly, to avoid delaying and lengthening this judgment.
648. It will help this approach to explain the overall dynamic of the arguments.
649. Claims 1 and 10 of EP565 can be considered to have a functional feature. The functional feature is the requirement in claim 1 for the formulated mRNA that is claimed to be a "vaccine", which means that it is able to generate an antigen-specific immune response. Claim 10 emphasises this requirement by stipulating that the vaccine is "for use in preventing and/or treating a BetaCoV disease", so it has to have a protective effect. The requirement of claim 10 is more stringent than that of claim 1 but for the purposes of the novelty and added matter arguments it does not seem to me to matter, both because of the way the parties argued the case and because Pfizer/BioNTech needs to knock out both claims.
650. I therefore refer below to "the functional feature" although in argument reference was also made to "functional features" because of this difference between claims 1 and 10.
651. All the other features of claims 1 and 10 are what Moderna described as "non-functional". I would prefer to describe them as "physical", but again it does not matter.
652. All that being so, the issue for novelty is whether the functional feature and the physical features of the claim are all, together, clearly and unambiguously disclosed by WO674, and the issue for added matter is whether they are all, together, clearly and unambiguously disclosed in the application as filed. So the test is materially identical (I refer back to what I said on the applicable law) but the starting point is different.
653. It is against this background that Pfizer/BioNTech positioned a squeeze between added matter and novelty. They said that if all the features are disclosed by the application as filed so as to avoid added matter then, applying the same standard in a consistent way, they must be disclosed by WO674.
654. Moderna did not deal with this squeeze directly in its opening written submissions. It denied anticipation and said there was no added matter, but did not address how both conclusions could be reached at the same time and applying the same standard. However, in its closing written submissions it committed itself in the following way (at paragraph 171, footnote omitted):
171. So far as the other non-functional features of claims 1 and 10 are concerned, on the bases that: (a) the approach in English law of 'clear and unambiguous' is the same for added matter; and (b) on the basis that the disclosure of the EP'565 application as filed is such as to provide support for claims 1 and 10 without adding matter, Moderna do not propose to take the point in this jurisdiction that the other non-functional features of the claimed composition relied on by Pfizer/BioNTech are not present in WO'674 for the purpose of novelty. Accordingly, the alleged squeeze between added matter and novelty does not arise.
655. This followed a section in which it had argued that there was no disclosure of the functional feature because Example 20 of WO674 is merely a proposal, relying on a line of cases including Hospira v Genentech [2014] EWHC 1094 (Pat), to which I return below.
656. So Moderna essentially sought, by concession, to say that both WO674 and the application as filed disclosed all the physical features of the claims of EP565, but that only the application as filed contained adequate disclosure of the functional feature, with WO674 falling short because of Example 20 being merely a proposal (by contrast, it said, the application as filed had repeated teaching about use of the mRNA formulations as vaccines, and data in the parts equivalent to Examples 23 and 24 of EP565, which are discussed above).
657. Moderna coupled this with the confident assertion in its written closing submissions that in fact the application as filed contained a better, i.e. clearer support for the claims than in WO674.
658. Pfizer/BioNTech struck back at this attempt to break up the squeeze in two main ways.
659. First, they maintained their case that there was added matter in any event, including in relation to the combination of the physical features. They said that the support in the application as filed was worse than in WO674.
660. Second, they said that on the assumption that all the physical features were disclosed in both the application as filed and in WO674, Moderna could not escape the squeeze by reliance on the functional feature because if a mere proposal was not enough for novelty then nor could it be for added matter. Pfizer/BioNTech said that a mere proposal that was plausible, even if unaccompanied by data, was enough to anticipate, and that when it came to added matter in this scenario Moderna would, because of the way that it was trying to evade the squeeze, need to show that the application as filed not only said that the physical features made the claimed products suitable for use as a vaccine, but made that plausible across the scope of the claims. Pfizer/BioNTech coupled this with detailed analysis of the Hospira line of cases and in particular the decision of Birss J (as he then was) in Merck v Ono [2015] EWHC 2973 (Pat).
661. In turn, Moderna said that Pfizer/BioNTech's introduction into the case of plausibility in the context of added matter was unpleaded and that its introduction would be unfair.
662. This way in which the arguments unfolded was therefore very intricate, but by explaining how they related to each other I can give my decision quite shortly.
663. First, the fact that Moderna "conceded" that both WO674 and the application as filed adequately disclose all the physical features of the claim does not necessarily make it so. I should consider whether it actually is the case, both for novelty and for added matter.
664. Second, I conclude, for reasons stated briefly below, that the application as filed plainly does not adequately disclose all the physical features together, so EP565 is invalid for added matter.
665. Third, I conclude that WO674 does not adequately disclose all the physical features together either, although its disclosure is, as Pfizer/BioNTech submitted, better than the application as filed. That means there is no anticipation in any event.
666. Fourth, I conclude that Pfizer/BioNTech's attempt to introduce an attack involving the plausibility of the application as filed is not pleaded and ought not to be allowed.
667. Before going on to give my more detailed reasons, I must digress to deal with an unfortunate procedural muddle.
668. During my reading for the start of the trial, the parties fell into disagreement about which document should be used to assess added matter. Pfizer/BioNTech in their opening written submissions used a document filed with the EPO on 20 March 2020, but Moderna said that that document had type that was too small to be legible and that I should for practical reasons therefore use PCT/US2016/058327, published as WO 2017/070626A, instead.
669. The former document is in fact the divisional application for EP565 ("the Divisional Application") and the latter is the parent application from which it derives ("the Parent Application").
670. Pfizer/BioNTech disagreed about legibility and anyway maintained that the Divisional Application was the legally relevant document. Moderna had not disputed that, and indeed in its written opening had said that the Parent Application should be used for the purely practical reason of legibility, and that the right comparison was with the Divisional Application. Moderna also actively submitted that there was no material difference between the documents.
671. I agreed with Pfizer/BioNTech about which document to use, for both the reasons given, and Pfizer/BioNTech opened the case by reference to it.
672. In fact, however, the Parent Application was the legally relevant document, not the Divisional Application. So Pfizer/BioNTech were wrong on that point, and Moderna had shared the mistake or at least not corrected it. I think the greater share of the blame lies with Pfizer/BioNTech since they were making the attack and had deployed the contention that the Divisional Application was legally the right document. I regret that I did not check the matter myself, but in my defence there were a great many issues to attend to and I took the parties at their word.
673. Although neither side had said there was any relevant difference between the two documents as to their contents, the Divisional Application in fact contains claims, not in the Parent Application, which would have advantaged Moderna had they been available as basis for EP565's claims. Pfizer/BioNTech dealt with those claims in its oral opening submissions without appreciating that they ought not to form part of the analysis.
674. Pfizer/BioNTech only noticed their error shortly before oral closings; their Counsel very properly apologised, and thereafter the mistake need not have mattered any more save for some inconvenience in correcting references, but Moderna objected that it was no longer open to Pfizer/BioNTech to argue based on the Parent Application, because it (Moderna) had relied on the error. I rejected that submission on the basis that it would be absurd for the Court to proceed on the wrong legal basis, and because all Moderna needed to do was to remove from its submissions any reliance on the claims of the Divisional Application. I did, however, allow Moderna time to rejig its submissions, because of the squeezes in play and the need for it to coordinate its argument across the two Patents and between the two leading Counsel. That meant an adjournment and the end of the trial was delayed by a number of days. As matters turned out Moderna's submissions changed very little and I do not think, with hindsight, that the adjournment was really needed.
Adequate disclosure of the physical features in the application as filed (added matter)
675. In the course of oral closing submissions I asked Counsel for Moderna what was its best or primary case on arriving at the physical features of the claims of EP565 from the passages relied on. His answer was that the "first half of the claim", concerning the betacoronavirus and the immunogenic protein came from pages 8 to 9 of the application as filed, from Examples 20 and 21, and from claims 43, 44, 79 and 80. He said that the LNP features came from page 90 line 29 and page 95 lines 6-11. He also relied on the fact that Prof Alabi had said (it was argued) that the composition of the LNP was "entirely standard" as a matter of CGK. And he said that the requirement that the structural lipid be cholesterol was disclosed because every time the structural lipid was specifically identified in the specification, it was cholesterol. He also relied on Examples 23 and 24. He said that Example 20 brought everything together bar the fact that the components of the LNP formulation were given as specific percentages and not ranges.
676. There are many fatal problems with this approach. Principal ones (they are interrelated) leading to my conclusion of added matter are:
677. First, it involves combining different parts of the disclosure when there is no teaching to do so. Counsel for Moderna said that it was enough that the facets relied on were disclosed as being preferred, but that is not so for many of them, even if one assumes that e.g. the scope of a dependent claim is an adequate indication of preference.
678. Second, it involves taking more specific teaching and using it to support broader features in the claims of EP565. For example, Example 20 not only has percentages for the LNP components where the claims of EP565 have ranges (itself a major stumbling block to which I will return in a moment), but also specifies precisely the cationic lipid, non-cationic lipid and PEG lipid. So Moderna's approach involves cherry-picking: taking the percentages but not the actual ingredients.
679. Then, as to the fact that Example 20 has specific percentages where the claims of EP565 have ranges, Counsel for Moderna sought first to say that the difference was unimportant (which is obviously wrong, since the introduction of ranges teaches the reader something new) and then tried to rely on Prof Alabi's evidence referred to above. But Prof Alabi was, as Counsel for Pfizer/BioNTech submitted, just saying that there were compositions that would fall within the ranges claimed that were obvious. He was not saying that the ranges were well known. Even if he were, that would be Moderna seeking, illegitimately, to use CGK not to understand but to supplement the disclosure of the application.
680. Third, it involves taking something relatively broader and trying to limit it by reference to just part of another, separate piece of teaching. For example, the claims of the Parent Application relied on (43 and 44) relate to a variety of antigenic polypeptides which includes the spike protein or S1/S2 and also the E, N and M proteins. But the claims of EP565 are limited to the S protein and S1/S2, a narrower choice not disclosed by claims 43 and 44. Moderna's answer to this was to rely on Examples 23 and 24. But those are specifically about MERS-CoV (and anyway are either full-length S or S2 so do not teach S1, or immunogenic fragments). Taking the broader list from claims 43 and 44 and then limiting it down with just the teaching in Examples 23 and 24 about the protein and ignoring the fact that they concern MERS-CoV presents the reader with new information: a new combination of features.
681. Fourth, it involves taking pieces of information and treating them as free-standing items of teaching to be plugged in anywhere else, at will, without a direction to do so. This was most apparent in relation to the formulation teachings. There are many different formulation percentages and ranges given, but they say nothing about which mRNAs encoding which proteins for which pathogens they might be used with, and I accept Pfizer/BioNTech's submission that the skilled person would think that the choice of protein could affect the appropriate formulation to use. They are not independent.
682. Fifth, it involves a mish-mash of features which are expressed to be preferred and ones which are not, or may seem less preferred. For example, claim 83 states precise percentages of the LNP formulation components. On Moderna's case this ought to be regarded as a statement of specific preference, but it does not match claim 1 of EP565, so instead Moderna focuses on claim 82 which names the components but does not give any percentages (specific or ranges). And Moderna's case expressly involves taking statements of preference in the form of claims and combining with teachings, especially about the percentages, which are just in a laundry list.
The Opposition Division Decision
683. The Opposition Division found EP565 invalid for added matter by a decision of 7 December 2023. Moderna submitted that it was a mechanistic decision in the spirit of what, it said, was an old-fashioned and unduly strict approach to added matter from which the EPO has moved away. I disagree. I think that the decision is a normal and straightforward application of up to date case law. I also think it is strongly consistent with my own decision, albeit that there are minor differences which may well be down to the arguments being presented differently. In particular I think that section 4.3.6, which says that there is no disclosure in the application as filed of the combination of the necessary LNP composition with a BetaCoV antigenic protein is in keeping with my analysis above; likewise section 4.3.8 focusing on the selection of S protein or S1 or S2 subunit.
684. The Opposition Division also found there to be added matter in the selection of cholesterol, a point which I have not spent time on above, because I think it is a relatively minor part of the picture. I do however agree with the Opposition Division and that would be another reason to find added matter.
Plausibility as part of the added matter argument
685. The relevant part of Pfizer/BioNTech's pleading is as follows:
Added Matter
3B. The matter disclosed in the specification of EP 565 extends beyond that disclosed in the EP 565 Application.
PARTICULARS
(a) Sub-paragraph 3A(b)(i) above setting out the features of claim 1 of EP 565 is repeated.
(b) The above combination of features is not clearly and unambiguously disclosed in the EP 565 Application and/or would require the impermissible selection from multiple independent lists in the EP 565 Application, and there is no clear pointer towards such a combination. Without prejudice to the foregoing, there is no disclosure of the combination of features (A) and (B) with (C) and (D) including, in the case of feature (D), where the final component selected is cholesterol as opposed to another structural lipid.
(c) Further or alternatively, there is no clear and unambiguous disclosure in the EP 565 Application of the combination of features (A), (B), (C) and (D) with the additional technical features of claims 2, 6, 7 and/or 10 of EP 565 and/or the same would require the selection from multiple independent lists without a clear pointer to such a combination.
3C. In relation to EP 565 as proposed to be amended conditionally, the Claimants will rely on the matters set out in paragraph 6 of the Statement of Objection. Further or alternatively, there is no clear and unambiguous disclosure in the EP 565 Application of the combination of the features of any of proposed conditionally amended claims 1A, 1B, 1C or 1D of EP 565 with the additional technical features of claims 4, 5 and/or 8 of EP 565 and/or the same would require the selection from multiple independent lists without a clear pointer to such a combination.
686. Paragraph 3A(b)(i), to which this refers back, is as follows:
3A.
(a) ...
(b) Without prejudice to the foregoing, the Claimants will say that claim 1 of EP 565 (and each proposed conditionally amended version thereof), and each claim dependent thereon, is not entitled to claim priority from any of priority documents P1 to P9 inter alia because:
(i) Claim 1 of EP 565 is to:
A. a BetaCoV mRNA vaccine comprising at least one mRNA polynucleotide having an open reading frame encoding at least one BetaCoV antigenic polypeptide,
B. wherein the at least one BetaCoV antigenic polypeptide is (a) a spike (S) protein or immunogenic fragment thereof; or (b) an S1 subunit or an S2 subunit of S protein or an immunogenic fragment thereof;
C. wherein the BetaCoV vaccine is formulated in a lipid nanoparticle,
D. wherein the lipid nanoparticle comprises 40-60% cationic lipid, 5-15% non-cationic lipid, 1-2% PEG lipid, and 30-50% cholesterol.
687. Thus these are explicit about the physical features and also mention "vaccine". Claim 10 is mentioned in addition at 3B(c). But there is no reference to plausibility of the vaccine effect, or treatment/prevention.
688. In paragraphs 215 to 216 of its written opening submissions, Pfizer/BioNTech said this:
215. Claim 1 discloses a vaccine which, in summary, both (1) comprises an mRNA polynucleotide encoding a betaCoV spike protein or S1 or S2 subunit or immunogenic fragment thereof; and (2) is formulated in an LNP comprising 40-60% cationic lipid, 5-15% non-cationic lipid, 1-2% PEG lipid and 30-50% cholesterol.
216. The Application discloses (1) a number of different possibilities for the
polynucleotides that can be encoded by the RNA; and (2) a number of different possibilities for the LNP formulation.
689. This was clearly focused on disclosure of the physical features, collected into (1) and (2). The word "vaccine" is used, but there is no suggestion that if the physical features were adequately disclosed together there would be an additional point about whether they would have a vaccine effect.
690. Moderna's opening written submissions denied added matter but also said that there was no anticipation of a treatment effect by a mere proposal, as I have mentioned above.
691. In oral opening submissions, Counsel for Pfizer/BioNTech floated the notion that this presented an additional squeeze point on added matter. Moderna had picked this up in time for its closing written submissions, in which it said (and I agree) that what was being argued was not entirely clear.
692. In its closing written submissions, Pfizer/BioNTech provided an Annex A which collated its arguments on anticipation by WO674 and on added matter into a chart. At this stage, however, it split the claim into three parts, in which (ii) and (iii) corresponded to (1) and (2) in its opening submissions, and (i) split out the vaccine requirement separately. There was now also an assertion that the application as filed did not disclose that something having the features from (ii) and (iii) would function as a vaccine. As matters developed in the written and oral closings, it became apparent that Pfizer/BioNTech was arguing both that plausibility should form part of the assessment, and that it was plain that examples 23 and 24 of EP565 did not render a vaccine effect plausible, at least not across the scope of the claims.
693. Pfizer/BioNTech answered Moderna's objection that this was an unpleaded and new point by saying that the "vaccine" feature was mentioned in its pleadings referred to above, and that the need to invoke plausibility had arisen in response to Moderna's reliance on the Hospira line of cases, also referred to above. I do not accept either of these points.
694. As to the first, the pleading is about disclosure and says nothing at all about plausibility, even contingently. It gave Moderna no clue that Pfizer/BioNTech might say that even if all the physical features were disclosed in combination, some additional point on "vaccine" might arise. I think it was reasonable for Moderna to think that if it could show an adequate disclosure of all the physical features together then that would be enough. After all, the application as filed is laden with disclosure that the goal of the products claimed is a vaccine; the added matter problem is all about definition of those products.
695. As to the second, I think it was entirely predictable that Moderna would rely on the Hospira cases to say that a mere proposal did not anticipate, and that Pfizer/BioNTech would need an answer. This whole side of the case is all about a squeeze that Pfizer/BioNTech was setting up, and it could and should have seen the issue coming if it wanted to run it.
696. I also think that if this had been flagged better and/or earlier then the evidence might well have been different. Certainly there was no exploration of plausibility across the scope of the claims based on Examples 23 and 24.
697. I therefore conclude it would be unfair to allow Pfizer/BioNTech to run this additional point, but it does not need it because EP565 is both obvious over WO674 (see below) and bad for added matter arising from the points on the physical features.
698. Since I do not allow the point in at all I do not need to go into the tricky question of the interrelationship between novelty and added matter when it comes to functional features of this kind and the consistency or otherwise of the Hospira line of cases and Merck v Ono. I am glad about that, not only because it avoids lengthening this judgment and the time to complete it, but because the argument took place in a rushed and unsatisfactory way at the very end of the trial and I would not be confident that I had had all the help I needed to decide it.
Adequate disclosure of the physical features in WO674 (novelty)
699. Pfizer/BioNTech's case was that there was a disclosure of all the features of claim 1 of EP565 in WO674 in the following way:
700. It starts with Example 20. That discloses an mRNA (for use as a vaccine) encoding a betacoronavirus S protein (in fact it is a MERS-CoV, although that is not necessary to get to the claims of EP565). Such is not in dispute.
701. Then Pfizer/BioNTech relies on the passage at page 866 that "Polynucleotides used in the studies herein which encode certain infectious agents antigens or variants thereof may be formulated in any of the formulations described herein including LNPs." This is very general, a point to which I will return in a moment. It also is embedded in a discussion of flu, and I do not think it is apparent that "certain infectious agents" means every infectious agent.
702. Lastly, Pfizer/BioNTech points to LNP formulations in the examples specifying a cationic lipid, a non-cationic lipid, a PEG, and cholesterol in the molar ratio 50:10:1.5:38.5. Those percentages fall within the ranges of claim 1 of EP565.
703. On the last point, Pfizer/BioNTech relies on the fact that there are no other LNP formulations disclosed in the examples. However, there are other formulations disclosed in the general teaching, specified by references to ranges which are not identical to claim 1 of EP565, although the range for each component generally overlaps. Pfizer/BioNTech did not submit that the formulations given in the general teaching disclose the relevant features of the claims of EP565.
704. It may also be noted that none of the examples where the formulation is given in detail involves MERS. Pfizer/BioNTech's point that the protein chosen and the formulation used interact, which it took on added matter, rebounds on it here.
705. This presents a more cogent picture than Moderna's case on added matter for EP565, but the problem is that it still requires choices to combine selected, different parts of the teachings in specific ways which are not taught. There are a variety of pathogens and proteins disclosed and a variety of formulations. Example 20 gives no formulation information at all. I do not think that the passage at page 866 says that every formulation applies to every infectious agent, and even if it did, the exercise would still involve choosing from a number of possibilities in respect of the agent and the formulation. I think it is unrealistic to position this as an individualised disclosure of every combination, which was effectively Pfizer/BioNTech's case.
706. So I conclude that Pfizer/BioNTech is wrong about the disclosure of WO674 being clear and unambiguous as to a combination of all the physical features.
707. Neither side structured its submissions explicitly by reference to the Pozzoli approach. I have set out my findings about the skilled team and the CGK above, and they have important implications for the obviousness case as I will explain in a moment. Those findings provide the input to the initial stages of the Pozzoli approach.
708. Given that the main attack is from Example 20 of WO674, the Pozzoli approach of identifying the inventive concept of EP565 and the relevant differences between it and WO674 would not work all that well anyway. The reason is that the differences on the face of WO674 and the claims of EP565 are not what the obviousness argument is about: Example 20 does not give any formulation information and so a number of the physical features of the claims of EP565 are missing and would be Pozzoli differences, but Moderna did not say that that is where invention lies. It effectively accepted that if (which it disputes) the skilled person decided to go ahead with an mRNA vaccine for MERS-CoV starting from Example 20 then there would be no invention in the details of the LNP formulation (in any event even if Moderna did not accept this, the evidence of Prof Alabi to that effect was unanswered), and the spike protein is expressly taught.
709. Rather, Moderna argued that the skilled team would not pick MERS-CoV to work on at all in the context of there being many other possible vaccine targets to progress, and would not have adequate expectations of success if they did think about it.
710. In outline, Pfizer/BioNTech's case was that the skilled team would read and understand Example 20 in the context of WO674, see it as an attractive way forward given that MERS was an important vaccine target (as was SARS), would know and understand that the spike protein meant there ought in principle to be neutralising antibodies, and would regard the LNP formulations given as a good platform which ought to work. They would have positive and certainly reasonable expectations of success even if there was no guarantee, based on the CGK and on the disclosure in WO674 itself of the positive results for flu.
711. Also in outline, Moderna's response was that WO674 is not really about coronaviruses at all, that they would not be on the radar of a NAV as vaccine targets at all, that the skilled team would not include know-how about coronaviruses, that coronaviruses were not significant vaccine targets of interest anyway, and that as a result of all those matters the skilled team would not take Example 20 forward but would regard it as an unimportant tailpiece and would focus on more important targets, more clearly flagged and supported by WO674 instead, for example flu. Even if the skilled person did think of taking Example 20 forward they would not have any reasonable expectation of success.
712. Many of these points are already in effect covered by my findings on the skilled team and CGK. In particular, Moderna's central argument that the skilled team would not have SARS/MERS on its radar fails in view of my findings on those topics. I will however go on to cover the parties' submissions in the light of those findings.
713. Moderna organised its submissions by reference to the list of factors in ICOS v Actavis, and for convenience I will address the points in that order (the ninth, "bonus effect", is not relevant). As ever, some are more important and more complex than others.
714. As I have already said, matters have to be assessed against the background of the teaching of WO674, which expressly teaches a vaccine against MERS being "a key vaccine", and my findings on the CGK, both that coronaviruses were important vaccine targets to which a rapid response might be needed, and as to the immune response (issues (a) and (e) on the CGK as addressed above).
715. First, prospects of success. Moderna argued that the prospects of success were absent, most strenuously on the twin, closely connected bases that Pfizer/BioNTech had led no evidence from a properly or adequately qualified expert and that neither Dr Ulmer nor Dr Sola had accepted that there was a reasonable prospect of success.
716. Given my findings that although Prof Dougan was a problematic witness his qualifications were adequate, and that his evidence was reliable as supported by documentary evidence on important points, I reject the all-or-nothing argument that Pfizer/BioNTech had no evidence for its case. Further, the objective facts are that many teams were working on coronavirus vaccines (albeit on different platforms), that the spike protein was an attractive basis for action and indicated that successful vaccination was likely to be possible for these viruses, and that very good results for flu were shown in WO674 (I note that Moderna positively argued that WO674 provided "proof of concept" for the LNP "platform", albeit in the context of flu). These facts were either not disputed or not capable of serious dispute.
717. Against that, the evidence of Drs Ulmer and Sola was in each case not to the effect that there was any definite reason to expect failure, but merely that nothing was certain until tested. Further, I do not think it is a fair and complete characterisation to say that they did not accept that there was a reasonable prospect of success. Given their inherently exceptionally cautious outlooks they were never going to say that, whatever the facts and whatever the project. But they did say there was reason for optimism and that was, for them, at the upper end of the positivity spectrum. See for example Dr Ulmer at T8/1145-1146 and 1254, Dr Sola at T9/1384-1385, 1389-1390.
718. Second, Moderna said that "such novel vaccine research" was "truly ground-breaking and cutting-edge work", hence not following an established path of research. This engages essentially the same points as in relation to prospects of success. Some of the research, in the sense of identifying the target pathogen and its immunological characteristics (the S protein in particular) had already been done and was well-established, given my findings on the CGK and the teaching of WO674. The LNP approach was somewhat new but that must not be overstated given the good results with flu in WO674, the established nucleic acid vaccine approaches, and the CGK Geall paper.
719. Third, burden and cost. There are a number of sub-points here. Moderna relied on Dr Ulmer's unchallenged evidence that the cost of bringing a new vaccine to market was about $500 million at around the EP565 Priority Date. That makes sense but is not, I think, a very relevant question. Any vaccine, however routine, would cost a huge amount. Any vaccine, however routine, could go wrong at a late stage in clinical trials for a large number of reasons inherent to any pharmaceutical venture and unrelated to the issues going to obviousness over WO674.
720. In addition, the main anxiety about prospects of success which Moderna levelled at WO674 was that the S protein would not be properly expressed or folded in the LNP platform or would not be protective. On the evidence I think the skilled team would expect such problems to be discovered at the animal model stage: still expensive but far short of the total cost of a vaccine development exercise.
721. Then, Moderna relied on the fact that the costs of pursuing WO674 were not just financial, but included the "opportunity cost" for the skilled team of not working on another vaccine target. Moderna said, with the support of Dr Ulmer who gave an example from his own experience, that this was an ethical issue because of the impact on the availability of a vaccine for that other target. As Moderna recognised, this interplays with the number of avenues of research available and I deal with it there.
722. Fourth, number of decisions and value judgments for the skilled team to make. Moderna said that there were multiple value judgments to make along the way, again relying on the evidence of Dr Ulmer. I disagree with this, at least to the extent that I reject it as a distinct point on top of the other matters that I am addressing in this section of my judgment. Of course, there would be a huge multiplicity of decisions and value judgments to make in any vaccine project, but that cannot make them all inventive to undertake. What I must concern myself with is value judgments at the level of the claims of EP565 and which bear on the decision of what if anything was obvious to do from WO674. WO674 expressly proposes a MERS vaccine based on the S protein in Example 20, and in the context of WO674 that would plainly be based on an LNP platform. The decisions necessary to land within the claims of EP565 are routine ones as to the precise formulation where Moderna did not say there was any invention and where in any case Prof Alabi was the only qualified witness, the decision to progress MERS rather than some other target, and the decision whether the endeavour had reasonable prospects of success. The choice of MERS and prospects of success arise under other of the Actavis v ICOS headings, which is why I say that this is not a distinct point in any event.
723. Fifth, alternative paths. Moderna argued that there were multiple other pathogens to seek to address, and multiple other vaccine platforms. This is probably the most complex point; the facts are essentially set out in my analysis of the disputed CGK, above. As I have said when addressing the law, the Brugger v Medicaid point ought not to be overdone; one ought not just to say that route X is obvious and therefore it does not matter what other routes there are. The existence of all those other routes can be a factor in assessing the obviousness of route X in the first place.
724. The global and industry background to this point is perhaps an unusual one. There was a very substantial degree of international cooperation and a desire to ensure that key pathogens did not go unaddressed. This was the concern both of industry and of international organisations such as the WHO. At the same time, however, even the major vaccine companies could each only afford to work on a relatively small number of targets.
725. In my view, there is some minor value in this point for Moderna, but not nearly as much as it argued for. Its arguments were so extreme that they would have led to the conclusion that the only obvious things to do in this field were to target the one or two most menacing pathogens, using only the most established techniques. This is, however, clearly not the reality of the situation at the EP565 Priority Date, with many companies working on numerous pathogens using a variety of platforms.
726. In addition, Moderna's reliance on other platforms being available is unconvincing given that WO674 is very clearly directed to mRNA vaccines using LNP formulations. The choice among pathogens is one that confronts the skilled team reading WO674, but the array of other platforms is not, unless they were considering rejecting WO674 altogether, a much harder argument to make and to which I give little credence. In dealing with the CGK I have noted that the work ongoing on coronavirus vaccines (see Modjarrad 2016) was in fact all using established platforms, but that there was no CGK attitude that that had to remain so.
727. Sixth, motivation. This focuses on the degree to which SARS and MERS were attractive targets for vaccine development. I have addressed this in relation to the CGK. There were very strong reasons to think that a vaccine for them would be desirable, and MERS at least was still highly topical given the outbreak in Korea in 2015. It is true that if one were to focus on any individual vaccine company in the real world in 2015, with capacity to work on only a couple of targets, they might well have more motivation to work on some other target(s), but that is really a different question. Having cautioned myself above about pushing Brugger v Medicaid too far, I nonetheless reach the firm conclusion that progressing a vaccine against SARS/MERS was an obvious thing to do, even though there were other obvious targets in the world.
728. Seventh, Moderna argued that the high level of efficacy of the claimed mRNA vaccines of EP565 could not have been expected, and in particular that Pfizer/BioNTech had no evidence that the high level of efficacy actually achieved by Moderna and Pfizer/BioNTech with vaccines against COVID-19 could have been predicted. Of course, Pfizer/BioNTech did not rely on success against COVID-19; that would have been the grossest hindsight and Moderna would rightly have objected to it. This point had no legitimate separate weight over and above the question of whether there were adequate prospects of success at the EP565 Priority Date based on CGK and WO674 and I have dealt with that above.
729. Eighth, Moderna warned against hindsight on the basis of (i) the Covid-19 pandemic and the efficaciousness of the mRNA vaccines of the parties, and (ii) its criticisms of Prof Dougan. I have borne both of these in mind.
730. Two points made by Pfizer/BioNTech are intertwined in the above but bear separate emphasis. First, the mRNA/LNP approach was not merely suggested by WO674 and an available approach, but had a specific advantage for viruses with pandemic potential (such as SARS and MERS), which was that it enabled rapid changes to be made to the antigen encoded. Second the S protein was a very attractive target and this point is not just a shield against the assertion that coronaviruses would be thought likely to fail or at the back of the queue; it was a positive reason to choose them to work on. These points go together positively to support Pfizer/BioNTech's case.
731. The ninth factor, whether a feature of a claimed invention is an added benefit in a context in which the claimed innovation is obvious for another purpose, was not relevant.
732. Taking all these together, I reach the clear conclusion that EP565 is obvious over WO674. Example 20 gives a clear pointer towards a goal that would be attractive, offering very good prospects of an effective vaccine, using a platform with attractive features that showed good results against flu and thus "proof of concept", against an important and well known target. The availability of other targets and platforms would be known to the skilled team but does not undermine my conclusion.
733. Moderna confirmed during closing oral submissions that it did not assert independent inventiveness for claims 6 or 7 if it lost on claim 1, so no separate point arises on them.
734. Pfizer/BioNTech only maintained Pardi weakly in the event that the obviousness attack from WO674 failed.
735. Pardi has no focus on vaccines and has no equivalent to Example 20 of WO674. This makes it a much weaker attack in general. Essentially the only scenario in which Pfizer/BioNTech said that it could play a part was if the WO674 obviousness attack failed on the basis that the results from flu were so good that that was the only obvious way to go.
736. This may be theoretically conceivable but it is not practically real. In any event, that is not the view I have taken of WO674. So Pardi does not add anything.
737. My views that EP565 is obvious over WO674 and that Pardi cannot in practical terms be any better if WO674 had failed are sufficiently clear that I am not going to burden this judgment with any further analysis of the obviousness case over Pardi.
738. I have set out the claims in issue above. Moderna did not maintain claims 6 and 7 for any purpose (these were finally, conclusively dropped only during oral closing submissions), but there still remain claims 2 and 10 and the proposed amended claims. It would have been better if Moderna had brought more discipline to this, earlier.
739. Given my conclusions about obviousness and added matter of EP565, I propose to deal with the dependent claims only briefly, directing myself principally to whether any of them can improve Moderna's position on those issues.
740. I have held that WO674 does not anticipate EP565 so Moderna would not need any dependent claims for that attack. In the (I think rather improbable) scenario that my decisions on added matter, obviousness and anticipation of claims 1 and 10 are all overturned on appeal, then the Court of Appeal would have to consider dependent and proposed amended claims, but I do not believe there is any question of fact that I need to resolve that would arise even then. I will however make brief findings on points of law about conditionally amended claims 1B and 1C.
741. Claim 2: this limits the encoded protein to an S protein. Moderna did not submit that this helped with obviousness and given my reasoning on that issue, I agree. It also does not help with added matter; in practical terms it could only make that problem worse, if anything, since it is to a still more specific selection. Claim 2 is for these purposes the same as claim 1A of the conditionally amended claims.
742. Conditionally amended claims 1B and 1C: these both contain a limitation designed to avoid anticipation by WO674, had that been necessary, by disclaiming the protein disclosed in Example 20 of WO674. The disclaimer is phrased by reference to the GenBank Accession Number. Pfizer/BioNTech argued that the proposed amendment does not fall within the limited categories of situation where an undisclosed disclaimer can be appropriate (novelty only citations, accidental anticipations, etc.). I agree with this. Therefore, Pfizer/BioNTech said, the disclaimer could only be allowed if it were disclosed, in the sense that the scope of the wide claim minus the matter disclaimed was disclosed in the application as filed, which is not the case. I agree with this too. Moderna never disputed these propositions. So these claims would not be permissible in any event, as they would add matter. Claim 1B also contains the same limitation to the S protein as does claim 2 and claim 1A and my comments above apply.
743. Conditionally amended claim 1D excludes non-self-replicating mRNA. This has no relevance to anticipation by WO674 so I do not need to deal with it. It could not improve Moderna's position on added matter anyway, and was not said to be relevant to obviousness.
744. Claim 10 does raise some additional issues, but I have dealt with them above, already.
745. As with EP949, there was a shepherding squeeze insufficiency plea (and a related AgrEvo obviousness plea) but Pfizer/BioNTech said in its opening skeleton that the pleas had done their job, so there is no need to address them any further.
746. My conclusions are:
i) EP949 is valid.
ii) EP949 is infringed, given that Pfizer/BioNTech conceded that it would be infringed if valid.
iii) EP565 is obvious over WO674.
iv) EP565 is not anticipated by WO674.
v) EP565 is invalid for added matter.
vi) None of the proposed claim amendments to EP565 makes any difference to these conclusions.
747. I will hear Counsel as to the form of Order if it cannot be agreed. I direct that time for seeking permission to appeal shall not run until after the hearing on the form of Order (or the making of such Order if it is agreed). I draw attention to paragraph 19.1 of the Patents Court Guide, which says that a hearing on the form of Order should take place within 28 days of hand down. In the present case, however, 28 days from hand down will be too close to the end of term so I direct that the hearing is to be in September.